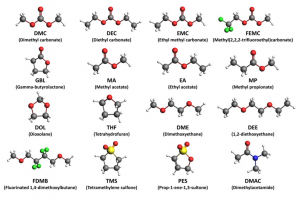
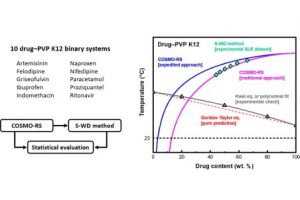
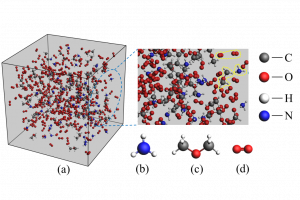
摘要 开发能够在极端条件下运行的锂金属电池电解质是一项重大挑战,并且往往因缺乏系统的溶剂筛选研究而受到阻碍。在这项研究中,通过 DFT 和 COSMO-RS 计算,来评估包含20种溶剂的190种二元混合物,以识别具有宽液体温度范围和高LiTFSI溶解度的电解质。四亚甲基砜(TMS)因其高沸点和低熔融焓而成为一种有前景的候选者,这会提高混合物中的泡点并降低共晶温度。利用具有七个σ描述符的机器学习模型,精确预测了Li和TFSI离子结合能。这些结合能主要受到强静电和范德华相互作用的影响。这种综合方法突出了DFT、COSMO-RS和机器学习技术相结合在指导电解质设计方面的有效性。 参考文献 Rational electrolyte design for Li-metal batteries operated under extreme conditions: a combined DFT, COSMO-RS, and machine learning study, J. Mater. Chem. A, 2024, 12, 15792