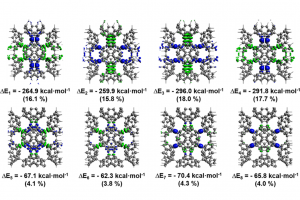
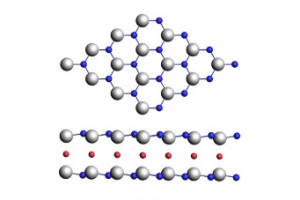
AdNDP轨道、超原子分子轨道(SAMOs)和NOCV轨道作用揭示二维富勒烯单层的电子结构和稳定性 研究背景 自从富勒烯C60被合成以来,富勒烯及其衍生物展现出令人瞩目的化学和物理性质。富勒烯是一种具有Ih对称性的二十面体结构,由20个六边形和12个五边形组成。这种独特的几何结构使它们与传统的纯碳材料如二维的石墨烯和三维的金刚石等材料截然不同。除了单个富勒烯分子外,以C60富勒烯为基本结构单元组装而成的各种碳材料也相继被合成。由于富勒烯封闭的笼状二十面体结构,富勒烯分子也具有很高的稳定性。所以在富勒烯材料中,C60分子之间难以形成强的化学键,主要依靠分子间的范德华(van der Waals)相互作用。例如,面心立方富勒烯晶体(fcc C60)已在室温下被合成,C60质心之间的距离为10.02 Å,其主要作用力为较弱的范德华力。 近年来,具有分子间化学键的富勒烯材料也被科学家们陆续发现。例如,通过[2 + 2]环加成键连接的六方相富勒烯(rhombohedral phase),以及在高温高压下合成的四方相富勒烯(tetragonal phase)等富勒烯固体材料。此外,研究表明,碱金属和碱土金属掺杂可以促进C60聚合物晶体的形成。这些掺杂的富勒烯聚合物展现出丰富的电子性能,有的甚至表现出超导现象。 最近,中科院化学所郑健课题组通过有机阳离子切片策略成功合成了二维单层准六方相富勒烯(qHPC60)和单层准四方相富勒烯(qTPC60)。在二维六方相富勒烯单层中,每个富勒烯被6个相邻的富勒烯碳笼包围,碳笼之间通过碳-碳单键和[2+2]环加成键连接。这种单层富勒烯聚合物的带隙为1.6 eV,具有优异的热力学稳定性,同时表现出优异的光学各向异性、机械性能和热电特性等。此后,关于二维富勒烯材料的实验和理论研究陆续大量报道,但对于这种材料的电子结构与其稳定性的深入探究仍有待进一步明确。 论文详情 清华大学化学系胡憾石、李隽团队采用密度泛函理论方法,通过研究二维富勒烯单层的电子结构和化学键来探讨其稳定性机制。理论计算表明,二维富勒烯单层具有1.46 eV 的直接电子带隙,与实验值(1.6 eV)吻合。研究发现,富勒烯分子间碳-碳键的形成对促进C60之间的电荷流动起着关键作用,使得 C60球内形成双重π芳香性,从而稳定了二维骨架结构。此外,研究还发现二维富勒烯单层内一系列离域的超原子分子轨道(SAMO)。这些轨道表现出类似原子轨道的行为,并杂化形成具有σ/π成键和σ*/π*反键性质的近自由电子带。本研究为理解二维富勒烯单层的电子结构和稳定机制提供了新的见解,对基于SAMO的近自由电子带的二维富勒烯材料的设计和潜在应用提供了理论指导。 该研究成果以“Understanding the Electronic Structure and Chemical Bonding in the 2D Fullerene Monolayer”为题,发表于Inorganic Chemistry期刊。南方科技大学博士后赵晓昆为本文第一作者,清华大学化学系胡憾石和李隽为本文通讯作者。 研究内容 1,二维富勒烯材料的结构和稳定性探究 Figure 1.二维富勒烯材料的结构,(a)俯视图;(b)侧视图。C1,C1′和C2分别代表[2+2]环加成键中的碳原子;C3和C3′分别代表分子间碳-碳单键中的碳原子。 在二维富勒烯材料中,由于C60分子间碳-碳键的形成,导致其内部的C60分子结构发生了扭曲,从Ih对称性降低到了C2h对称性。同时,C60分子内的“56”键和“66”键长的范围分别为1.377-1.610 Å和1.358-1.505 Å;而在原始的C60分子中“56”键和“66”键长分别为1.452和1.397 Å。此外,C60分子间[2+2]环加成键(C1-C1′)和碳碳单键(C3-C3′)的长度均为1.606 Å。对该二维富勒烯材料内部的碳-碳键强度的分析表明,分子间碳-碳键的强度(~ -7.9 eV)要明显小于C60分子内部的碳-碳键强(> -11.2 eV)。对不同温度下(分别为600 K、1000 K、1400 K和1800 K)下时长30 ps 的AIMD模拟结果表明,该材料在1400 K以上仍具有良好的热力学稳定性。然而,在1800 K时,该材料在10 ps的AIMD模拟后会分解成单个的C60分子。 2,二维富勒烯材料的芳香性探究 […]