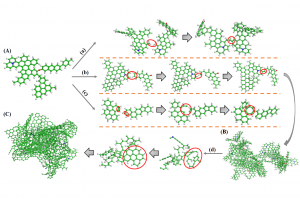
相关背景 煤沥青是煤焦油蒸馏工艺的副产品,由于其具有含碳量高、杂元素含量低、廉价且来源广泛等优势。对煤沥青进行改质可以获得更高的结焦值和软化点,改质煤沥青是石墨电极、碳纤维、碳纳米管、C/C复合材料等高附加值碳制品的重要优质原料。改质煤沥青的焦化是制备高附加值碳素产品的必经工序,它决定了产品的真密度、机械强度、电导率、石墨化度等性能。探索改质煤沥青焦化机理对优化高附加值碳材料的制备工艺具有重要意义。然而,现有科学文献对MCTP的微观结构组成及焦化过程的深度演化机制研究较少,本文精确构建了改质煤沥青的分子结构模型,并基于ReaxFF分子动力学方法对改质煤沥青的焦化机理在原子尺度上进行了深度研究,为实现改质煤沥青更高值化的利用,以及高附加值碳材料制备工艺的改进提供理论了依据。 研究亮点 通过一系列的检测分析(XRD, 13C NMR, FT-IR, XPS, MALDI-TOF-MS等)获得了MCTP的微观结构特征和组成。MCTP的碳骨架主要由芳香结构组成,芳香结构单元主要为苯和萘。脂肪族结构主要以甲基和亚甲基的形式存在。MCTP的分子量分布集中在1500 Da范围内。MCTP中含O、N和S的官能团分别以醚、季氮和无机硫的形式存在。在此基础上,建立了能够表征改质煤沥青微观结构特征和组成的分子模型(C93H59NO)。利用AMS2019软件中ADF模块计算改质煤沥青分子模型的量化性质,得到的13C NMR和FTIR图谱与实际检测结果吻合,验证了模型的合理性。 运用所建立的分子模型、ReaxFF MD模拟(AMS2019软件ReaxFF模块)、TG-MS、XRD和SAXS分析,揭示了改质煤沥青焦化过程中的挥发份去除及结焦机理。改质煤沥青焦化过程中挥发物的去除是由改质煤沥青分子边缘结构的破坏和活性自由基的产生引起的。主要挥发产物为H2、H2O、CO、CH4和C2H4。挥发分主要在430 ~ 900 K的温度范围内析出。对于焦化过程,XRD和SAXS分析表明,沥青焦样品的结晶和石墨化程度在挥发分去除阶段(低于973 K)被破坏,在高温阶段(>高于973 K)得到改善,Lc、Nc、La、Rg和孔隙率在1573 K时分别达到3.81 nm、12.03 nm、1.68 nm、18.54 Å和9.9%。在ReaxFF MD模拟过程中,沥青焦核的真密度(最终为2.2 g/cm3)、RDF临界峰强度、sp2杂化键比例(最终为41.4%)和六元环比例(最终为63.3%)均呈现先下降后上升的趋势。实际检测分析(XRD、SAXS)和ReaxFF MD模拟得到的结论可相互映证。沥青焦核的形成可分为两个阶段:焦核的形成阶段和有序化阶段。在形核阶段,改质煤沥青分子中的开环反应和脂肪链的交联导致了初级焦核的形成,主要通过三条反应路径,在路径一中,芳香烃通过芳香甲基直接连接;在路径二中,由边缘六元环断裂形成脂肪链,然后通过脂肪链结合实现芳烃的聚合;在路径三中,两个芳烃边缘的C原子相互结合形成五元或七元环结构。在有序化阶段,在初级焦核内会发生缩聚和芳构化反应,最终形成沥青焦微晶核。 图1 文章摘要图 图2 研究流程示意图 图3 改质煤沥青分子结构模型及验证 图4 改质煤沥青结焦机理图 参考文献 Zihan You, Jin Xiao, Gang Wang, Zhen Yao, Ye Wan, Qifan Zhong, Molecular representation and atomic-level coking evolution investigation of modified coal tar pitch […]