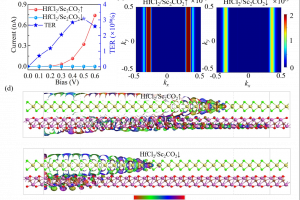
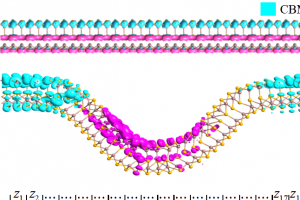
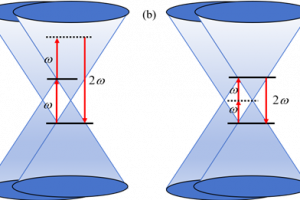
研究简介 谷电子学作为突破传统电子器件性能瓶颈的新范式,其核心挑战之一在于实现高效非易失的谷电子行为调控。结合密度泛函理论计算与非平衡格林函数模拟,福州大学材料科学与工程学院萨百晟课题组联合宁波东方理工大学(暂名)周通课题组,提出了一种HfCl2/Sc2CO2范德华异质结,实现了铁电调控的非易失谷电子行为。研究表明,单层Sc2CO2的铁电极化主导了HfCl2/Sc2CO2范德华异质结的电子结构特征:正极化会在能谷处诱导直接带隙,使异质结展现出谷电子行为,可通过圆偏振光进行读写;负极化则形成间接带隙,抑制谷电子行为。非平衡格林函数输运模拟计算进一步表明,基于HfCl2/Sc2CO2的铁电p-i-n隧道结的最大隧道电阻(TER)比率可达1.60×108%,揭示了其在低能耗谷电子与铁电非易失存储器件中具有良好的应用前景。福州大学材料科学与工程学院博士研究生崔舟为论文第一作者。该研究得到了国家重点研发计划与国家自然科学基金的资助支持。 研究内容 本文系统研究了单层HfCl2、Sc2CO2与HfCl2/Sc2CO2范德华异质结的电子结构、谷电子特性及其铁电隧道结的电子输运行为。图1展示了HfCl2和铁电Sc2CO2单层的相关性质,其中单层HfCl2和Sc2CO2均为间接带隙半导体,且单层HfCl2的价带顶(VBM)和单层Sc2CO2的导带底(CBM)处均由于自旋轨道耦合(SOC)效应产生了谷自旋分裂。 图1 单层HfCl2和Sc2CO2的(a)晶体结构、(b)静电势(Ep)和功函数(WF),(c)HfCl2和(d)Sc2CO2单层不考虑(w/o-SOC)和考虑(w-SOC)自旋轨道耦合时的能带结构。 图2展示了HfCl2/Sc2CO2异质结的相关性质,正极化的HfCl2/Sc2CO2↑和负极化的HfCl2/Sc2CO2↓异质结分别表现出具有0.22 eV直接带隙和1.01 eV间接带隙的半导体特性,且HfCl2/Sc2CO2↑异质结中的谷电子学能带结构特征得以保持,能够通过特定圆偏振光进行读取。而在HfCl2/Sc2CO2↓异质结中,谷电子能带结构特征消失。由于两种极化性质的HfCl2/Sc2CO2异质结的带隙大小和谷电子行为差异显著,在异质结中可同时实现谷电子与铁电存储机制,如图3所示。图4进一步展示了基于HfCl2/Sc2CO2↑异质结的Berry曲率,阐述了异质结中可能存在的光学跃迁选择规则和谷霍尔效应。 图2 (a)HfCl2/Sc2CO2↑和HfCl2/Sc2CO2↓范德华异质结的晶体结构和极化翻转过程的动力学路径。(b)HfCl2/Sc2CO2↑和(c)HfCl2/Sc2CO2↓范德华异质结在不考虑自旋轨道耦合(w/o-SOC)和考虑自旋轨道耦合(w-SOC)情况下的投影能带结构。 图3 HfCl2/Sc2CO2范德华异质结的谷电子与铁电存储机制。 图4 HfCl2/Sc2CO2↑范德华异质结在(a)整个二维布里渊区和(b)沿着高对称点的Berry曲率,(c)自旋分辨的谷光学跃迁选择规则,(d)K和K’谷的谷霍尔效应示意图。 最后,构建了如图5所示的p-i-n铁电隧道结开展非平衡格林函数电子输运计算。发现在0.5 eV偏压下,沟道长度为6 nm的隧道结器件实现了3.09×106%的最大隧道电阻(TER)比率。当沟道长度增长至8 nm时,其TER比率可提升至1.60×108%。这些结果不仅为铁电调控谷电子跃迁提供了设计思路,同时也表明HfCl2/Sc2CO2范德华异质结是一种极具潜力的低能耗谷电子存储器与非易失存储器候选材料。 图5 基于HfCl2/Sc2CO2范德华异质结的p-i-n结的结构与输运特性。(a)HfCl2/Sc2CO2↑和HfCl2/Sc2CO2↓ p-i-n铁电隧道结的结构示意图,其中S和D分别表示源极和漏极。(b)有限偏压下的透射电流曲线及隧道电阻(TER)比率。(c)在零偏压下费米能级处的自旋依赖透射谱。(d)在E = 0 eV及(kx, ky)=(0.37, 0)处的器件输运本征态。 参考 Z. Cui, X. Duan, J. Wen, Z. Zhu, J. Zhang, J. Pei, C. Wen, T. Zhou, B. Wu and B. Sa, Ferroelectric control of valleytronic nonvolatile storage in HfCl2/Sc2CO2 heterostructure, Appl. Phys. Lett. 126, 122902 (2025), doi: 10.1063/5.0264472.