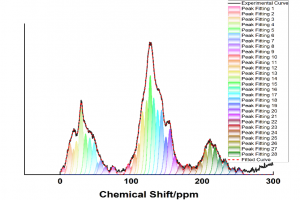
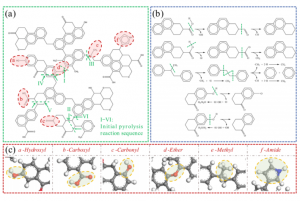
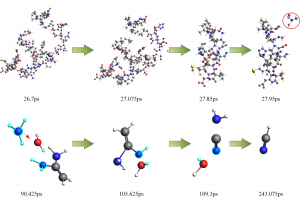
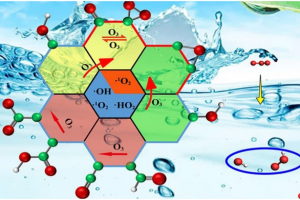
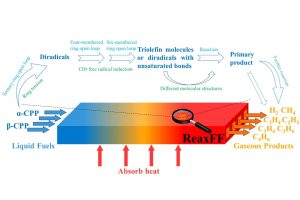
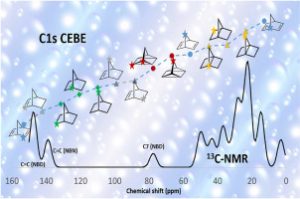
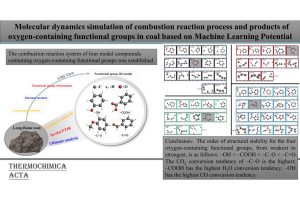
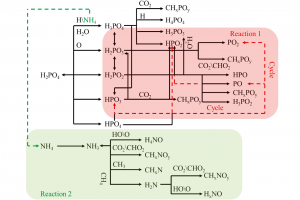
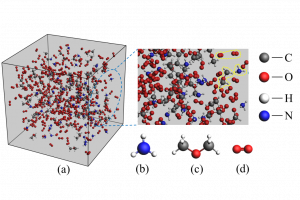
研究背景 褐煤作为一种低阶煤,有挥发分和水分含量高的特点,导致其热值较低且易发生自燃,严重影响着能演工业的储存、运输和利用。当前研究从宏观层面揭示了褐煤的燃烧机制,但其背后具体的分子级燃烧特性尚未被充分理解,特别是在不同高温条件下的反应路径与动力学行为仍缺乏系统研究。目前,对于褐煤在高温下燃烧行为的系统性数据匮乏,制约了其化学动力学机制的深入挖掘。因此,深入了解褐煤的燃烧特性与自燃特性,并在此基础上优化其燃烧过程,对于提高能源利用效率、减少环境污染具有重要的现实意义。本文以神木褐煤为对象,基于表征实验构建并优化其大分子结构模型,采用Amsterdam Modeling Suite(AMS)软件与ReaxFF MD方法,对其在不同高温条件下的燃烧过程进行了系统模拟。重点分析了不同温度下神木褐煤的总势能变化、燃烧产物特征及其高温燃烧机制。模拟结果验证了自由基在煤燃烧反应链中的关键作用,尤其在高温条件下,通过自由基的持续生成与消耗维持复杂的燃烧过程。目前该文于2025年4月6日以“Construction of macromolecular model of Shenmu lignite and study of its high-temperature combustion mechanism using ReaxFF MD simulation”为题在Fuel(中科院SCI二区 TOP)期刊上发表。第一作者为辽宁工程技术大学大学安全科学与工程学院贾进章教授,通讯作者为2022级硕士研究生田昊同学。本工作得到国家自然科学基金(No. 52174183,52374203)和辽宁省博士科研启动基金项目(No.2023-BS-203)的资助。 图文速览 图1 SM 褐煤的XPS光谱 图2. SM褐煤中元素氮和硫的峰值拟合光谱(a)氮;(b)硫 图3. SM褐煤样品的13C NMR分峰拟合光谱 图 4. SM褐煤的实验13C NMR光谱与模型计算13C NMR光谱的比较 图 5. SM褐煤大分子结构模型(a)优化前;(b)优化后 图6. 纯氧条件下SM褐煤的高温燃烧模拟 图7. 不同温度下褐煤势能随时间的变化情况 图8. 四种气态产物在不同温度下的时间演变(a)O2;(b)H2O;(c)CO2;(d)CO 图9. 三种自由基在不同温度下的时间演变 图 10. 不同温度下 SM 褐煤燃烧的反应总数变化 图11. […]