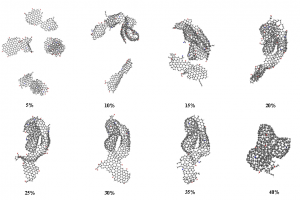
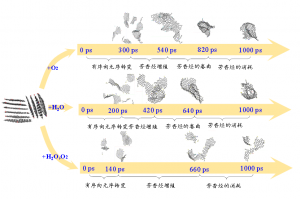
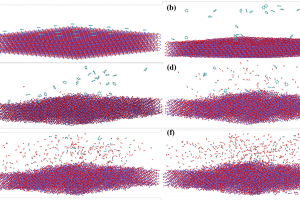
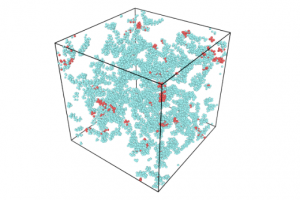
简介 含碳固体燃料(煤炭,生物质,石油焦,含碳废物)的精细化利用是未来能源发展趋势。水蒸气气化技术可以将固体燃料转化为更加清洁的气体燃料(CO,H2)。近年来,微波加热技术由于其高效和易控制特性而被引入气化技术中。除了微波热效应外,有学者发现微波对气化反应具有显著的非热效应。而交变电场对焦炭气化反应的化学层面的作用是微波非热效应机理的关键。燕山大学赵登课题组通过分子动力学模拟和量子化学计算探究了焦炭在电场作用下的气化机理。研究发现:电场的存在可以显著促进焦炭气化反应,电场的促进效果随电场强度的增大成指数增大。有意思的是,交变电场对气化的影响不仅仅来源于电场本身,电场的方向和强度变化会显著促进反应过程。同时对于气化的副反应,电场对水煤气反应和水解反应都有着明显的促进效果。 研究内容 本研究中所有的分子动力学模拟均使用AMS软件的ReaxFF模块,在C/H/O/N力场下进行模拟。从伊利诺伊州6号煤焦芳香烃片层模型中选取了的10个芳烃片层,建立初始的焦炭模型C1037H241N10O26,添加2800个H2O分子。为了将电场场强变化和方向变化分离开,设置了正弦、方波与恒强三种波形以及不加电场的空白对照组;同时设置了不同的电场强度,以研究场强大小对气化过程的影响。为了尽可能地将非热效应与热效应隔离开来,使用Berendsen控温器进行加电场的NVT系综的恒温分子动力学模拟。 图1 波形示意图(a)恒强(b)方波(c)正弦 图2 焦炭模型和水分子模型结构(C:灰色;H:白色;O:红色) 图3 不同场强作用下的气化反应过程的碳转化率变化 图3表示了3000K下不同波形体系在1ns内的碳转化率,图中可以直观的看到三种电场对气化的影响存在着差异,这体现出了微波自身的不同属性(场强大小变化,方向变化)对气化反应的影响不同。综合来看,在相同的温度下,电场的引入可以显著促进气化反应,这体现出了微波对于焦炭气化反应的非热效应,正弦电场对焦炭气化反应的促进作用是三种波形电场中最显著的。除了电场本身对气化有影响,电场的场强变化及方向变化都会对气化过程有促进作用。 图4 3000K下不同场强对气化反应过程的碳转化率的影响(a)正弦电场系统中碳转化率与电场强度的关系(b)方波电场体系(c)恒强电场体系(d)500ps下的碳转化率与电场场强的关系图 从图4(a)~(c)可以看出随着场强的增大,电场对气化反应过程的促进越明显。但是可以看出在0.01场强之前电场对气化反应过程促进作用较弱,超过0.15之后,电场的促进作用显著增大,这表明场强对气化的作用可能存在一个阈值。图4(d)显示,对于所有三种场类型(正弦波、方波和恒强电场),随着电场强度的增加碳转化率成指数增长。焦炭的微波气化过程中在电场强度达到一定程度之后,焦炭所受到的电场的影响程度会急剧升高,这可能是因为,焦炭碳边缘的碳氧复合物结构受场强的影响很大,随着场强变化芳香性破坏。 图5 反应过程中正弦电场体系与无电场体系中碳环数量的变化 对反应过程中的碳环数量进行了统计,如图5,可以看出:正弦电场体系中6元环的消耗要快于无电场体系,这证明电场的引入促使6元环破坏,形成不稳定的5元环与7元环,从而促进了气化过程。因此电场可以通过影响焦炭边缘的稳定性促进气化。环结构的稳定性与芳香性是相关联的。 图6 3000k下焦炭在不同电场作用下的水蒸气气化产物随时间变化分布图 在焦炭水蒸气气化过程中,气化剂产生的含氧自由基为O自由基与OH自由基,这两种自由基作为活性物质将O携带吸附到Char边缘。从图6中可以看出波形对于O自由基和OH自由基的生成影响不大。但加电场后对于OH自由基和O自由基的生成都有很明显的促进作用,无电场体系中没有搜索到O的生成,加电场后OH自由基的含量分别约为无电场体系4-5倍。 图7 无电场作用下的气化反应路径 图8 正弦电场作用下的气化反应路径 对无电场体系与正弦电场体系气化路径进行了分析。无电场体系与正弦电场体系对比,无电场体系中,CO的生成过程如图7,其大致为羟基吸附到活性位点然后析出CO,正弦电场体系中有较多O自由基直接吸附到C原子上并破坏碳环的现象,如图8中的A和C路径,因此O自由基的活动可能是正弦电场促进气化反应速率的一个因素。相同反应路径下,正弦电场的体系气化反应的脱附过程要比无电场的体系快10倍左右,例如图7中的B路径与图8中的B路径。 (a) (b)图9 3500K(a)无电场体系与(b)正弦电场体系中焦炭形态随碳转化率的演变图 图9(a)为无电场体系中焦炭形态随碳转化率的演变图,可以看到随着反应的进行焦炭的形态由松散逐渐变得紧凑,多数片层相连形成大的片层,其中存在片层堆叠的情况,当碳转化率到15%时出现了卷曲结构,在碳转化率为20%-40%期间这个卷曲结构进一步卷曲堆叠使结构越发紧凑,使裸露的边缘活性位碳位点减少,碳转化率到达到40%时形成球状结构,边缘碳活性位点进一步减少,气化反应是从焦炭边缘开始进行,而卷曲与堆叠行为在边缘位置减少的前提下,更多碳被包裹很难接触到外界水蒸气,反应会进一步变慢。图9(b)展示了正弦电场体系中焦炭形态随碳转化率的演变情况,在相同转化率下,焦炭结构在正弦电场体系中比无电场体系更松散,碳转化率由5%到35%并没有出现堆叠与卷曲现象,其主要的变化为片层相连形成二聚体或三聚体。因此电场引入通过对片层间的卷曲与堆叠行为的削弱影响气化反速率。 图10 Char模型范德华表面静电势分布(a)未施加电场(b)平行于Char平面强度为250 a.u.的电场(c~f)垂直于Char平面,电场强度依次为150,250,350,450 a.u.(1 a.u.≈ 51.423 V/ Å) 为了探究吸附过程中电场对于焦炭边缘电荷分布的影响,利用简化的焦炭模型的DFT计算获得的电子波函数,对波函数进行图形化处理得到焦炭边缘范德华表面的静电势分布如图10所示。无外加电场下,在氧原子处形成负电势,其余碳边缘上形成正电势。在垂直于Char平面方向施加电场后(图c),碳边缘正电势极值较无电场时由22.39kcal/mol增大至26.65kcal/mol,并且随着电场强度增大,逐步增加至66.61kcal/mol。边缘静电势的增大加强了对含氧自由基的吸附。在平行于Char方向施加电场使得焦炭平面两侧电子逆电场方向发生转移,产生静电势极化现象,在两侧形成较大的正/负静电势(-60.39kcal/mol,89.61kcal/mol),这远大于同场强下垂直方向电场的碳边缘静电势,这表明了沿焦炭平面方向的电场是促进气化吸附过程的主导因素。 图11 垂直于Char平面的不同电场下Char模型的定域化共轭π电子轨道等值面图(lol-π=0.3)(a)无电场(b)平行于Char平面强度为250 a.u.的电场(c~f)分别为垂直于Char平面强度依次为150,250,350,450a.u.的电场 图12 成环原子(C3,C4,C7,C8,C9,C10)的多中心键级及Char形态变化 简化模型焦炭分子平面具有共轭π电子轨道,通过分析共轭π电子轨道,可以直观显示焦炭芳香性的变化,芳香性决定了分子的稳定程度。图12可以看出,随着场强增大,多中心键级在150-250 a.u.电场场强下变化较小,达到350 a.u.场强之后开始减小,450 a.u.场强下大幅减小为负值,从图11(f)来看,450 a.u.场强下碳环上的π键完全破坏,导致了多中心键级的剧变。所以电场能够通过破坏焦炭边缘的芳香性来促进气化的脱附过程。另外,焦炭平面和碳氧复合物扭曲方向与电场方向相关,在正弦电场或方波电场中电场方向在不断改变,这造成焦炭分子会在不同方向上发生扭曲震动,焦炭边缘碳氧复合物会在这样的震动下不断被拉扯,从而更容易脱离焦炭边缘。 总结 本文通过AMS软件的ReaxFF模块对焦炭微波气化总体过程进行了等温模拟研究,并对气化过程进行分解,分别对吸附过程,脱附过程和水煤气反应进行了单独模拟研究来分析微波在各个反应中的作用。通过DFT的方法研究了电场对焦炭分子的影响。研究发现: 焦炭的微波气化过程要比非微波气化过程快8~10倍,电场的场强大小变化和方向变化对气化过程有明显的促进作用,且两者的作用可以叠加,相同场强下正弦电场对气化过程的促进效果最佳;电场强度对微波气化过程的影响成指数变化。电场主要通过两方面促进气化吸附过程,一方面促进O自由基和OH自由基的生成,进而促进了气化过程的吸附过程,另一方面电场使Char发生极化进一步促进吸附过程;电场可以显著加速脱附过程的进行,电场的方向变化对脱附过程也有可观的促进作用,电场的引入会使焦炭边缘的芳香性与多中心键级发生变化,达到一定的场强后芳香性被破坏,多中线键级突变,这解释了气化反应速率与场强的指数关系,此外电场对碳氧复合物有一个动力学的拉扯作用,这进一步促进了脱附过程的进行。电场的引入对于水煤气反应具有明显促进作用,且电场场强变化与场强大小变化都有明显作用,并且发现恒强电场对水的解离过程促进效果更佳。 参考文献 Jian S ,Deng Z ,Haoyuan F […]