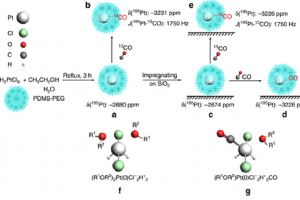
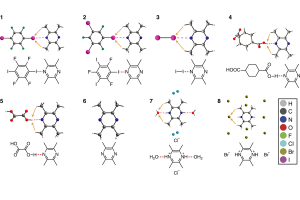
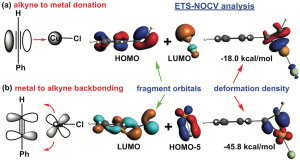
核磁共振(NMR)谱是阐明化学结构最重要的分析方法之一。然而对于某些原子核来说,可能的化学位移范围很大,相应的核磁共振测量往往很耗时。因此,利用稳健的量子化学方法进行可靠的化学位移预测具有广泛的意义。 为了评估用于预测119Sn NMR化学位移的常用方法和新量子化学方法的性能,最近一项研究提出了一个新的基准集,称为SnS51,包括50种含Sn化合物,其中Sn具有不同的键基序,分子大小从4到209个原子不等。共有51个119Sn NMR化学位移实验值,范围从2448到-2204 ppm,可作为计算方法评估的参考。 为了计算119Sn NMR位移,评估了15种密度泛函,将它们与三种不同相对论方法(标量X2C、ZORA和自旋轨道耦合ZORA)相结合,并基于CREST/CENSO算法生成的异构-旋转异构体集合,评估构象的柔性对化学位移预测的影响。此外还研究了119Sn NMR化学位移计算的结构依赖性,以及半经验量子力学(GFN2-xTB)或力场(GFN-FF)方法的适用性。为了进一步改善结果,作者研究了一种简单的线性标度方法。 研究表明,稳健的杂化泛函如PBE0,与TZP基组、COSMO溶剂化模型与自旋轨道耦合相对论哈密顿量相结合,总体平均绝对偏差良好,在100~ppm以下(另见NMR常见问题)。 参考 文献: J. B. Stückrath, T. Gasevic, M. Bursch, and S. Grimme, Benchmark Study on the Calculation of 119Sn NMR Chemical Shifts, Inorg. Chem. 2022, 61, 3903-3917