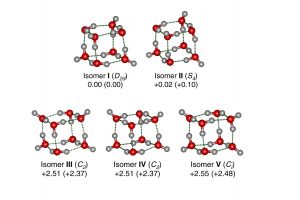
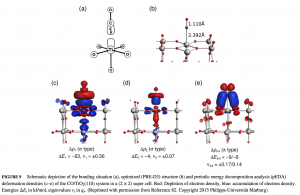
水八聚体的立方结构由六个四元环组成的团簇体系,能很好的用来解释氢键拓扑结构细微变化所驱动的协同作用。虽然许多不同的结构被预测出来,但从振动光谱中提取出结构信息仍待实现,这需要电中性团簇的尺寸选择性具有足够的分辨率来识别不同异构体的贡献。清华大学/南方科技大学李隽课题组、胡撼石课题组、中国科学院大连化学物理研究所杨学明课题组、张东辉课题组和江凌课题组报导了使用可调真空紫外自由电子激光器阈值光离子化方案,测得孤立的冷冻、电中性八聚水的特定尺寸红外光谱,结果观察到大量的尖锐振动带特征。 对红外光谱的理论分析表明存在五个立方异构体,其中两个具有手性。这些结构的相对能量反映出不同的拓扑相关、离域多中心氢键作用。这些结果表明,即使有共同的结构特征,氢键网络之间的合作程度差异也导致了不同层次的结构。 为了解水八聚体的电子结构,作者利用离域定域分子轨道(LMO)理论分析了立方异构体的氢键网络,进行了自然键轨道(NBO)、自适应自然密度分配(AdNDP)、能量分解分析-化学价自然轨道(EDA-NOCV)和主相互作用轨道(PIO)等分析。 利用AMS软件ADF模块,在GGA-PBE/TZ2P水平上,利用EDA-NOCV分析了立方异构体中氢键相互作用的的本质。EDA-NOCV方案提供了关于化学键中轨道相互作用强度和贡献的定性(Δρorb)和定量(ΔEorb)信息。 参考文献: Gang Li, Yang-Yang Zhang, Qinming Li, Chong Wang, Yong Yu, Bingbing Zhang, Han-Shi Hu, Weiqing Zhang, Dongxu Dai, Guorong Wu, Dong H. Zhang, Jun Li, Xueming Yang & Ling Jiang, Infrared spectroscopic study of hydrogen bonding topologies in the smallest ice cube, Nature Communications volume 11, Article number: 5449 (2020)