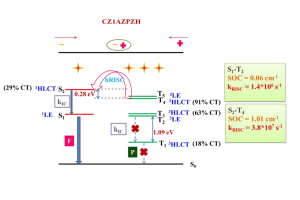
近年来TADF过程的研究取得了诸多突破,但要进行更高效率和量子产率的TADF分子设计,还需要更多深入的理论机理上的研究。与传统(冷)TADF一样,基于热激子的TADF材料也可以有效地利用单重态和三重态激子,理论上产生100%的IQE。与冷TADF(从低激发T1到S1)不同,热TADF中的RISC过程发生在高激发三、单重激发态(Tm(m>1)与Sn(n>1))之间。设计满足热激子形成条件的材料,例如低三重态之间能隙足够大,而高激发单-三重态能级间隙足够小,仍然是相当困难的。 印度SRM大学化学系Jesni M Jacob、Mahesh Kumar Ravva近期的研究中,探索、分析了分子设计的基本概念,并通过密度泛函理论建立热TADF分子的结构-性质关系,提出了一种分子设计策略。作者设计了一系列新的热激子机制施主(D)-π-受体(A)型分子,探索了新设计分子的电子特性,以助于设计“热激子”通道OLED材料。由于苯恶嗪(PXZ)和咔唑(CZ)的给电子能力适中,因此选择它们作为给电子单元,而吡嗪单元上的吸电子基团包括H、F和CN被取代为受体单元,使用CN化的萘噻二唑(NZ)和蒽噻二唑(AZ)单元连接供体和受体,设计出十二个D-π-A框架分子。这项研究可以为具有多个热激子通道的有机材料的分子设计方法带来新的见解,从而更好地利用激子。 参考文献 Theoretical Insights on Molecular Designing of Hot-Exciton based Thermally Activated Delayed Fluorescence Molecules, J. M. Jacob and M. K. Ravva, Mater. Adv., 2022, DOI: 10.1039/D2MA00039C