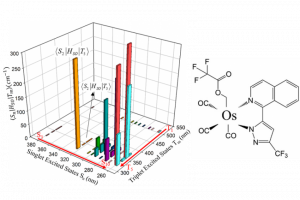
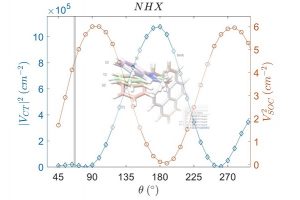
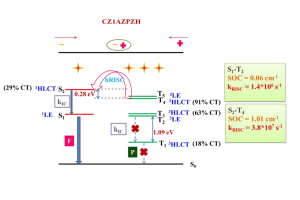
第二激发态三重态通过热激活反向内转换促进 TADF 和三重态-三重态湮灭光子上转换 近年来,由于 TADF 材料的高激子利用效率、低生物毒性等,TADF 有机发射体数量激增。然而现有的 TADF 通道在设计高性能材料方面还不够完善,特别是高位三重态介导的反向系间交叉过程。本文报道了一种热激活的反向内转换(TARIC)途径,该途径可以通过势能面锥形交点,实现从 T1 状态到 T2 状态的有效转移,势垒为 3.81 kcal mol−1。 在此基础上,介导的 T2 态促进了 TADF 和三重态-三重态湮灭光子上转换(TTA-UC)通道。此外,得益于TARIC途径,设计的 2′,7′-二氯荧光素(DCF)衍生物 DCF-MPYM-Me 光敏剂具有 13.6% 的优异上转换效率,高上转换效率对不含重原子的纯有机 TTA-UC 体系 TADF 光敏剂来说至关重要。 The Second Excited Triplet-State Facilitates TADF and Triplet–Triplet Annihilation Photon Upconversion via a Thermally Activated Reverse Internal Conversion, Yanliang Zhao, Yingnan Wu, Wenlong Chen, Ruiling Zhang, Gaobo Hong, Jiarui Tian, Honglei […]