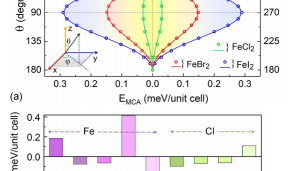
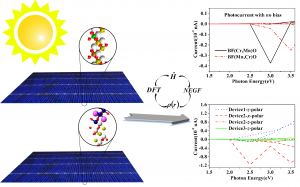
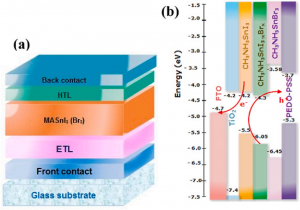
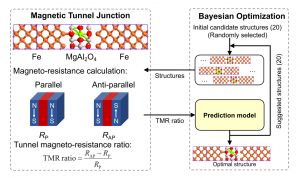
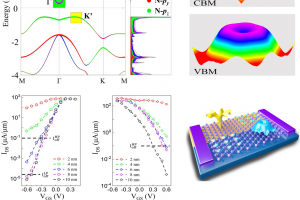
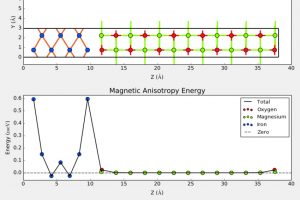
摘要 具有本征半金属性的超薄二维铁磁体在纳米自旋电子学器件设计中具有很好的应用前景。在这项工作中,作者利用密度泛函理论(DFT)系统地研究了一类有前途的二维铁磁体单层二卤化铁(FeX2,X=Cl,Br,I)的自旋输运和动力学性质。全部计算使用QuantumATK 完成,作者充分使用了 DFT 的 LCAO 基组、平面波基组方法,并在不同方法之间进行了对比。分析计算则包括自旋分辨能带、二维费米面、自旋极化的电子输运(DFT-NEGF)、声子计算、磁各向异性、Gibert Damping(Kambersky torque-torque关联模型)。 首先,作者使用非平衡格林函数(NEGF)与 DFT 结合方法,研究了 FeX2 单分子膜的自旋输运性质。研究显示了体系固有的半金属性和较大自旋带隙,在较宽费米窗口(>1ev)上可以产生 100% 的自旋极化。 在此基础上,作者深入研究了它们的磁各向异性、Gilbert 阻尼和交换相互作用,这是控制自旋动力学的关键。作者的模拟方法包括使用 force theorem 来确定磁各向异性,采用 Kambersky 的转矩-转矩关联模型确定 Gilbert 阻尼。计算结果揭示了这些材料中相当大的垂直各向异性(0.04到0.25 mJ/m2)以及相对较低的 Gilbert 阻尼(7.9×10−5 到 3.7×10−4)。 作者在计算模拟结果的基础上,利用自旋极化格林函数理论,探讨了这些材料中的有效交换相互作用,并研究了它们的自旋波刚度(spin-wave stiffness)、交换刚度常数(exchange stiffness constant)和居里温度(Curie temperature)。所有这些计算都提供了这些 2D FeX2 铁磁体作为下一代自旋电子学应用材料的重要性依据。 参考 原文:Ram Krishna Ghosh, Ashna Jose, and Geetu Kumari. Intrinsic spin-dynamical properties of two-dimensional half-metallic FeX2 […]