
文章摘要
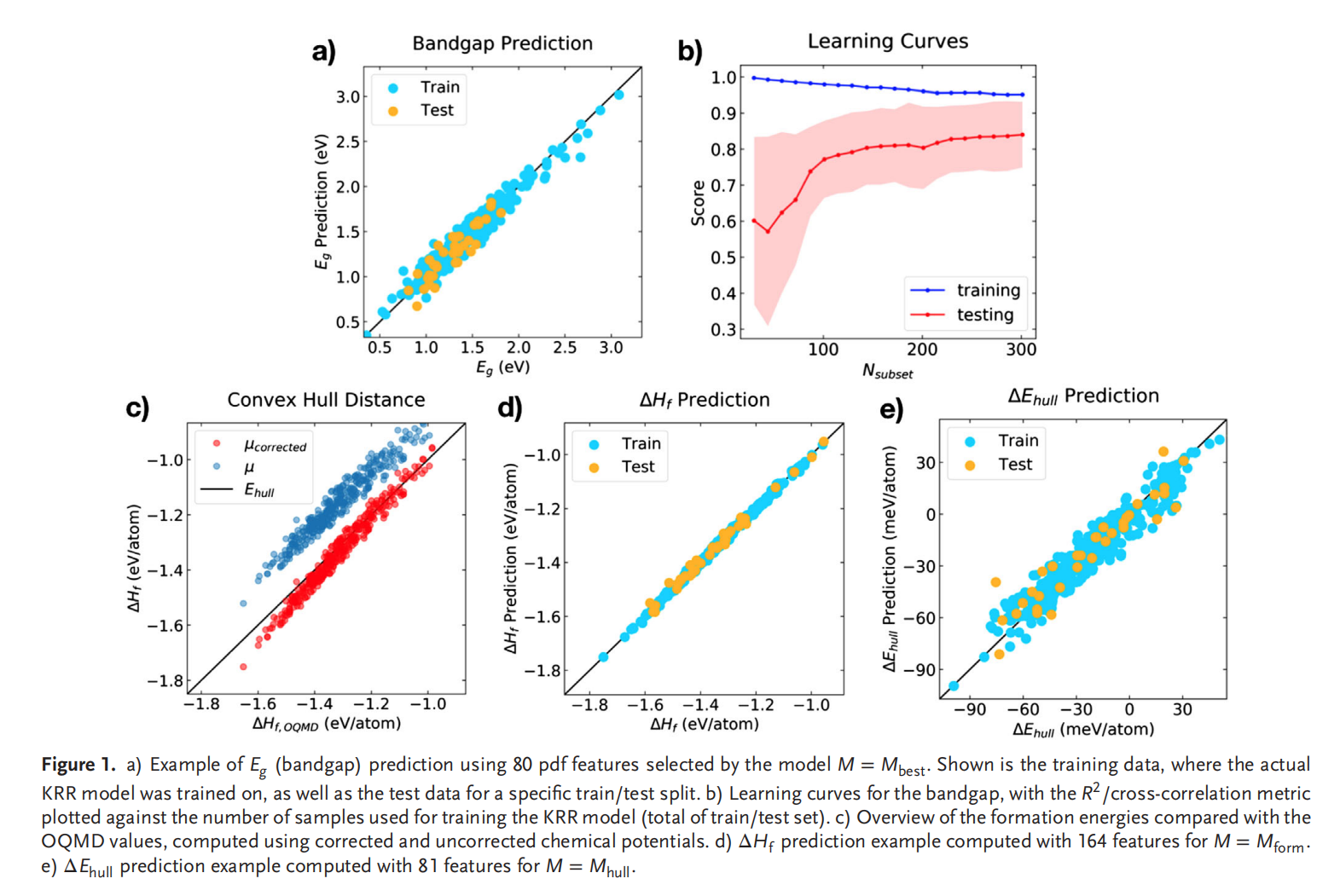
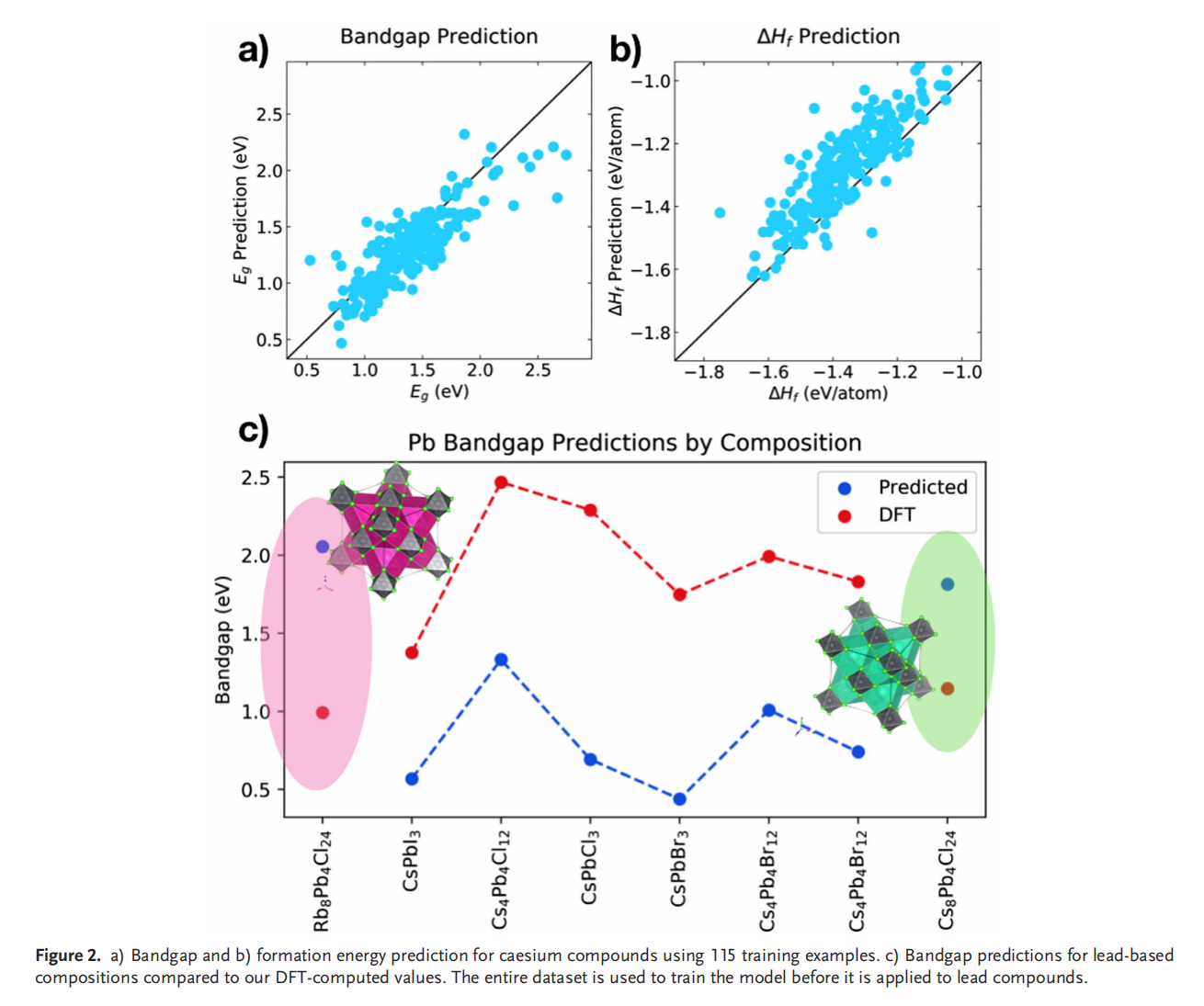
钙钛矿的成分调控让人们能够精确控制其在光伏应用所需的材料性能。然而,同时解决效率、稳定性和毒性仍然是很大的挑战。混合的无铅钙钛矿和无机钙钛矿最近显示出了解决此类问题的潜力,不过它们的组分空间巨大,即使采用高通量方法也很难发现有希望的候选结构。此项研究通过使用由密度泛函理论生成的344个钙钛矿的新数据库,使用与元素无关的通用指纹信息的机器学习方法可以快速而准确地预测关键属性。使用验证子集预测的带隙、形成能、和凸包距离分别在146 meV、15 meV/atom 和 11 meV/atom 内。得到的模型可以用于预测完全不同的化学组分空间中的趋势,并进行快速的组成和结构空间采样,而无需进行昂贵的从头算模拟。
QuantumATK中的计算自动化功能
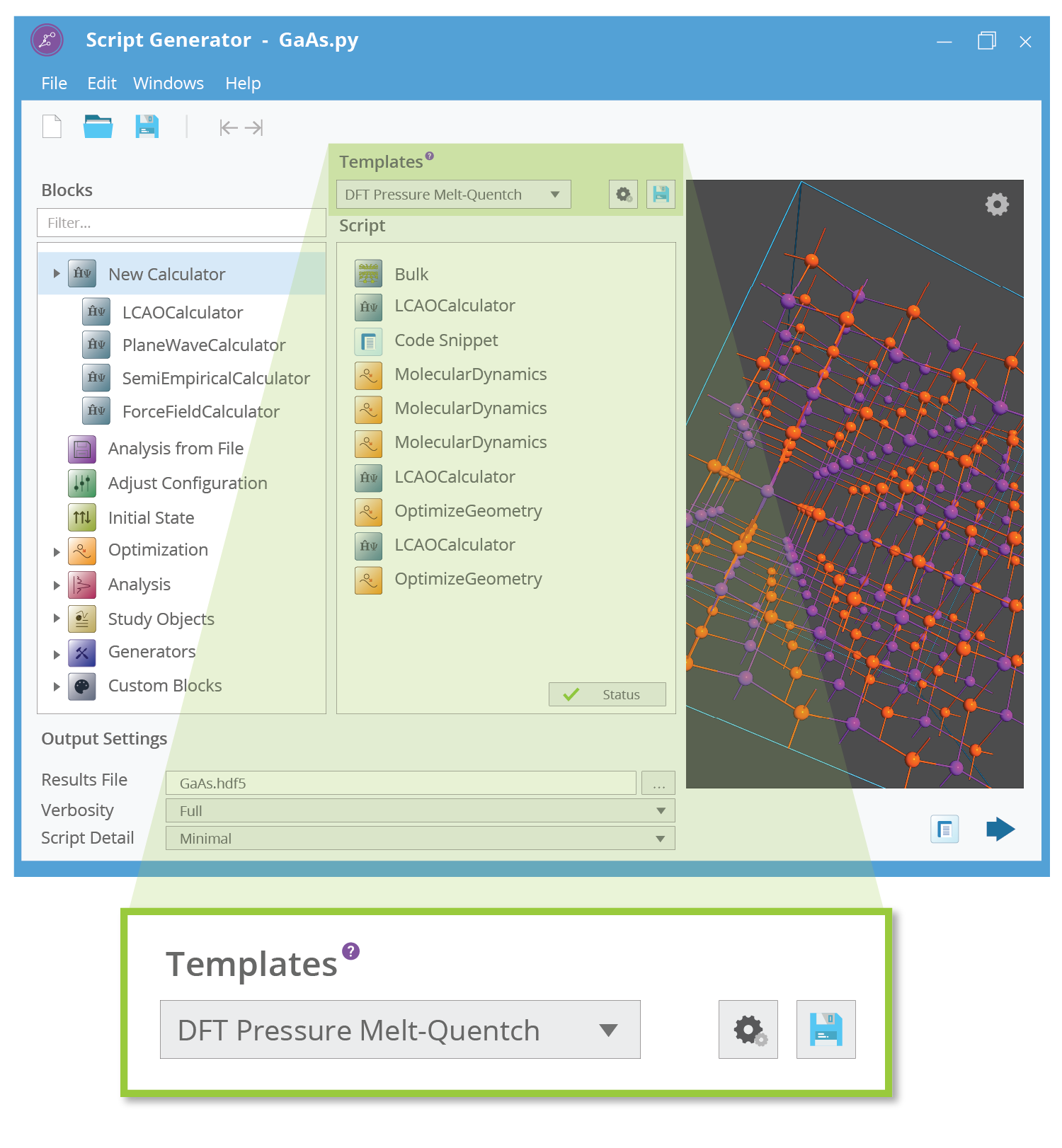
本文中提及的研究全部采用QuantumATK完成。QuantumATK包含了完整的Python 3基础环境与科学计算模块支持,还包含众多自主开发的结构生成、计算方法和性质分析模块,可以直接设计自动化的计算流程:
- 结构模型:使用 Python 脚本操作结构、设计结构模板、批量生成结构;在图形化界面的 Builder 中也可以使用 Python 命令操纵结构。参考:https://docs.quantumatk.com/guides/builder/builder_console_guide.html
- 计算设置:使用 Python 脚本自动化完成批量结构的计算;图形界面也可以直接设置计算脚本模板:
https://docs.quantumatk.com/guides/scripter/templates/templates.html
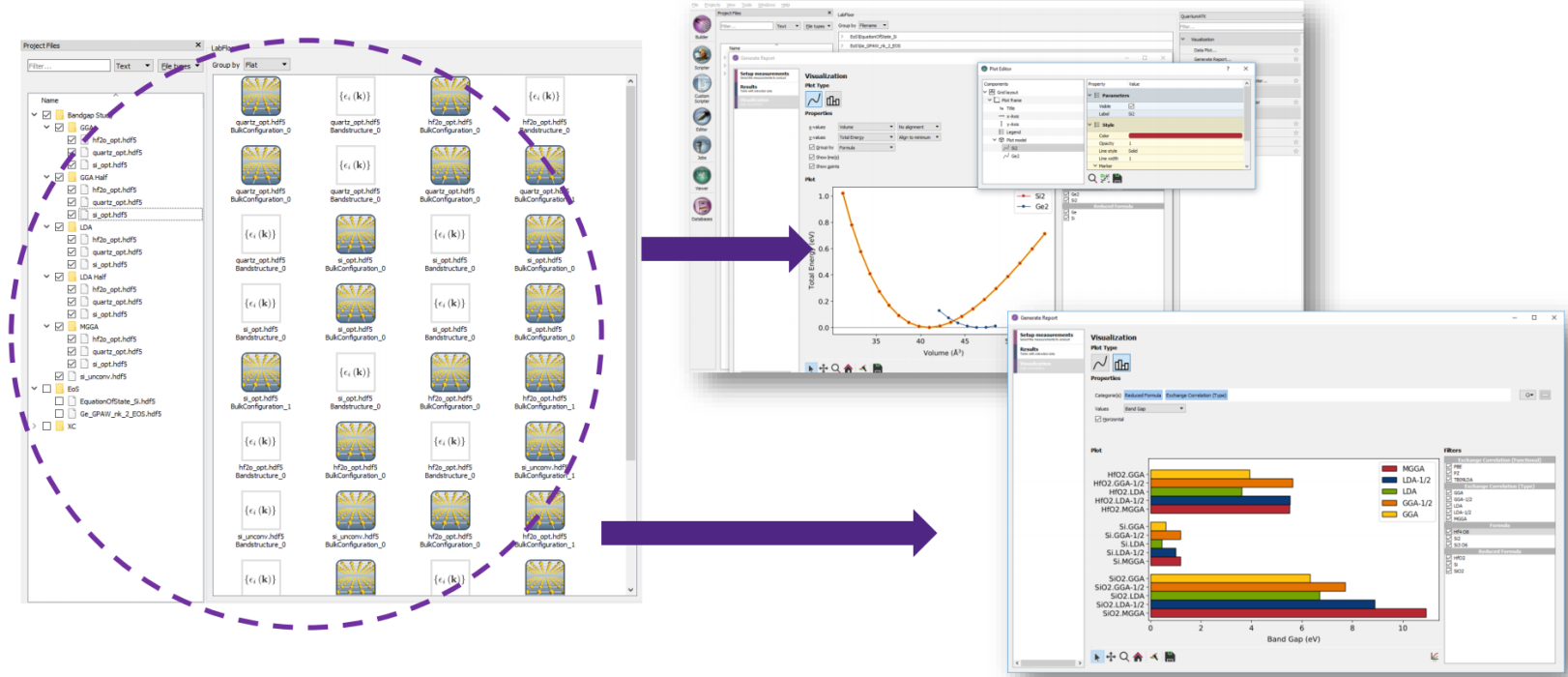
- 性质分析与汇总:图形用户界面新增报告生成工具,直接汇总计算结果得到统计和对比图。
参考
- Jared C. Stanley, Felix Mayr, and Alessio Gagliardi. Machine Learning Stability and Bandgaps of Lead-Free Perovskites for Photovoltaics. Adv. Theory Simul. 2019, 1900178
- QuantumATK脚本编程手册:https://docs.quantumatk.com/manual/NLRefMan.html
立即试用 QuantumATK!