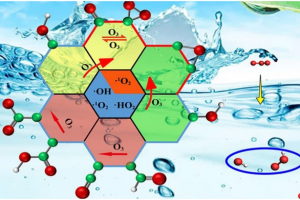
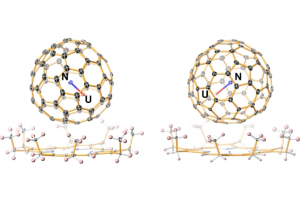
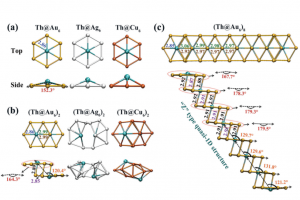
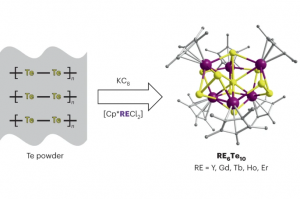
多氧钒酸盐中成分驱动的原型动力学 在计算质谱学中,人们经常需要解决将质谱信号与可行且化学上合理的结构相关联的问题。尽管这些问题在有机化学领域中通常能够得到解决,有时甚至使用简单的启发式方法,但对所有无机和纳米分子体系来说仍然是一个挑战,例如配位化合物会表现出多种键合连接方式。 在最近发表在《化学科学》上的一项工作中,剑桥大学Kondinski 等人解决了由钒、砷和氧原子制成的新的、复杂、多核无机物的质谱信号相关性问题。从组成上来说,无机分子属于球形杂多钒酸盐 (heteroPOV) 家族,这是一组众所周知的催化活性和磁响应材料。 确认质谱仪检测到的结构,最初似乎是一个无法克服的问题,正如作者在论文中仔细列举的那样,有超过 40,000 种构造可以在形式上构建出球形heteroPOV。作者决定采取不同的路线并应用系统思维,整合了关于已知结构的经验证据,并将分子内应变作为替代构型的一个因素,从而能够较快地在整个化学空间中搜寻,并迅速定位到一些可能的结构。使用 AMS软件中ADF模块优化、整理并分配,与实验观测相关联的分子结构。 总体的研究揭示了一种前所未有的结构变化:当球形heteroPOV逐渐被砷过度地取代时,整体结构将跃变为环状。 Kondinski, M. Rasmussen, S. Mangelsen, N. Pienack, V. Simjanoski, C. Nather, D. L. Stares, C. A. Schalley, W. Bensch, Composition-driven archetype dynamics in polyoxovanadates, Chem. Sci., 2022, 13, 6397-6412 离子性可控的超原子盐 弗吉尼亚联邦大学Turbasu Sengupta与Shiv N. Khanna发现一类新的金属-硫族超原子分子。以Rh6S8Ln为例,通过在金属-硫族团簇周围结合适当的配体,可以改变双超原子分子的成键特性。选择不同配体,与超原子中相同原子结合时,配体的电负性随种类而有所不同,作者选择了膦配体PR3,其中R官能团为NME2、F等,分别产生不同的电负性。作者发现,这项技术也能用于(PR3)5Rh6S8–Rh6S8(CO)5二聚体、盐的电子特性控制。两边电子能级的差异导致内部产生了电荷转移,从而在共价键基础上增加了离子键成分,而且导致这种二聚体产生了偶极矩/内部电场。也就是说,二聚体的偶极矩/内部场的强度可以通过改变PR3基团的电负性来调整。用这种偶极强度、内部场和HOMO/LUMO位置可调的超原子分子团簇组装成的固体材料,可能会有特别的用途,例如光捕获。 此外,还观察到这种超原子分子离子的氧化还原性质也可以使用类似方法得到预测和调制,即通过改变PR3基团的电负性实现。这种超原子分子也可以考虑用来组装成铁电固体,作者目前正在与其他实验课题组合作探索应用。 Superatomic salts with controlled ionicity, Turbasu Sengupta, Shiv N. Khanna, Mater. Adv., […]