
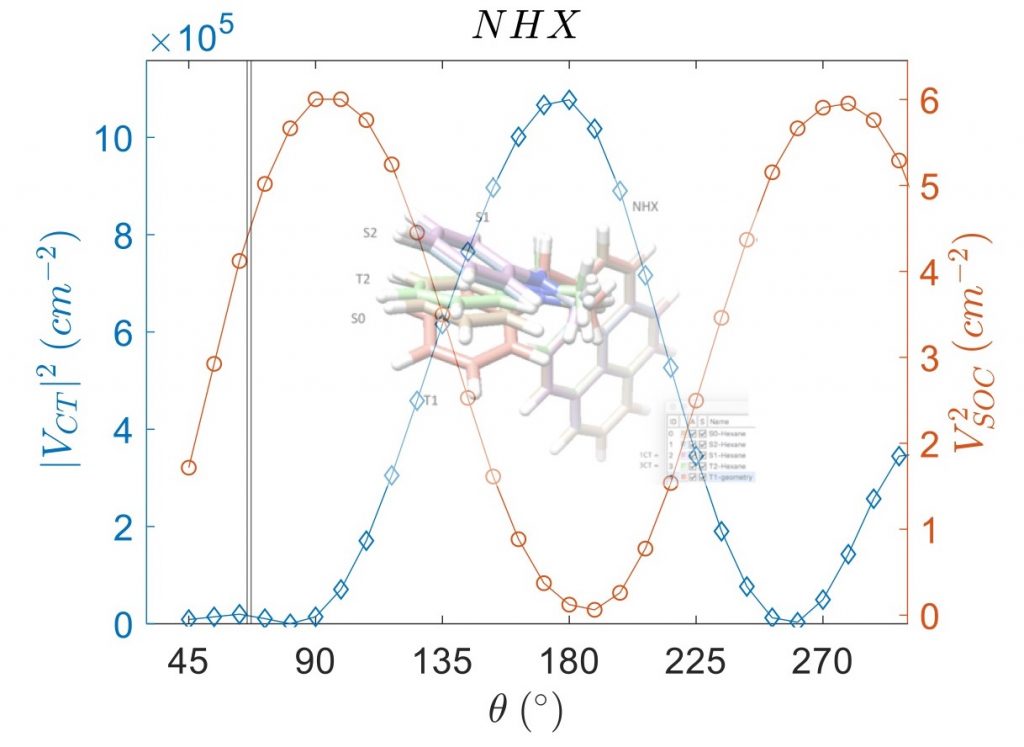
电荷转移态中的自旋轨道耦合
阿姆斯特丹大学的研究人员研究了电子供-受体分子的扭曲对性质的影响。对于包含芘受体和二甲基苯胺供体的电子供体-受体体系,确定了自旋轨道耦合矩阵元(SOCME)、电荷分离的电子耦合,对构象的依赖性。
对热激活延迟荧光(TADF)、光动力疗法、三重态发光二极管而言,自旋轨道耦合效应起着决定性影响。作者在动力学和能量角度,讨论了旋-轨电荷转移系间窜跃 (SOCT-ISC) 机制,包括经典Marcus电子转移理论中,电荷分离、电荷复合的相关参数。自旋轨道耦合,在电荷复合到三重态过程中起着重要作用,可以通过TD-DFT 进行探索,同时TD-DFT也为理解和预测 SOCT-ISC 机制提供了有效途径。该研究用丙酮和 4-硫代胸腺嘧啶的自旋轨道耦合矩阵元作为基准。
关于这项工作的三个报告的视频资料,可以辅助读者理解如何在自己的工作中使用类似的方法:
- 芘-二甲基苯胺正交电荷转移态的自旋-轨道耦合_René Williams
- 使用计算化学来描述和理解SOCT-ISC机制_Davita van Raamsdonk
- 正交电荷转移态中的自旋轨道耦合_Shivan Bissesar
所有 ADF 输入文件(链接)。
- Bissesar, D. M. E. van Raamsdonk, D. J. Gibbons, R. M. Williams, Spin Orbit Coupling in Orthogonal Charge Transfer States: (TD-)DFT of Pyrene-Dimethylaniline. Molecules 2022, 27 (3), 891.
热激子基 TADF 分子设计的理论探讨
近年来TADF过程的研究取得了诸多突破,但要进行更高效率和量子产率的TADF分子设计,还需要更多深入的理论机理上的研究。与传统(冷)TADF一样,基于热激子的TADF材料也可以有效地利用单重态和三重态激子,理论上产生100%的IQE。与冷TADF(从低激发T1到S1)不同,热TADF中的RISC过程发生在高激发三、单重激发态(Tm(m>1)与Sn(n>1))之间。设计满足热激子形成条件的材料,例如低三重态之间能隙足够大,而高激发单-三重态能级间隙足够小,仍然是相当困难的。
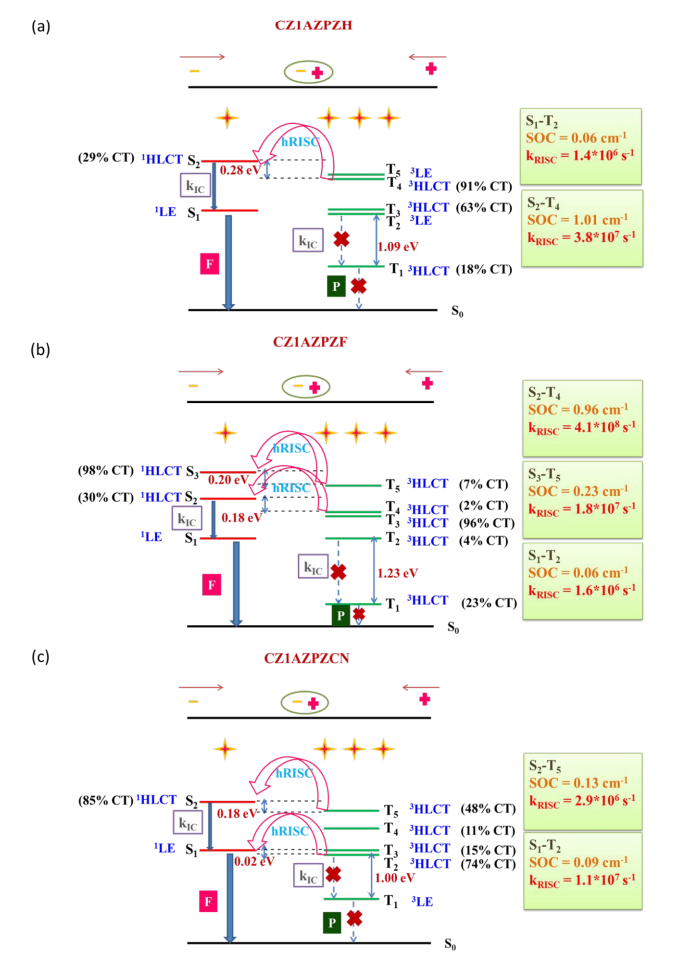
印度SRM大学化学系Jesni M Jacob、Mahesh Kumar Ravva近期的研究中,探索、分析了分子设计的基本概念,并通过密度泛函理论建立热TADF分子的结构-性质关系,提出了一种分子设计策略。作者设计了一系列新的热激子机制施主(D)-π-受体(A)型分子,探索了新设计分子的电子特性,以助于设计“热激子”通道OLED材料。由于苯恶嗪(PXZ)和咔唑(CZ)的给电子能力适中,因此选择它们作为给电子单元,而吡嗪单元上的吸电子基团包括H、F和CN被取代为受体单元,使用CN化的萘噻二唑(NZ)和蒽噻二唑(AZ)单元连接供体和受体,设计出十二个D-π-A框架分子。这项研究可以为具有多个热激子通道的有机材料的分子设计方法带来新的见解,从而更好地利用激子。
- Theoretical Insights on Molecular Designing of Hot-Exciton based Thermally Activated Delayed Fluorescence Molecules, J. M. Jacob and M. K. Ravva, Mater. Adv., 2022, DOI: 10.1039/D2MA00039C
基于高效 TADF 咔唑金树状聚合物制备出溶液加工型 OLED
香港大学化学系分子功能材料研究所,最近首次设计并合成了一类新的含C^C^N配体的咔唑金(III)树状大分子,其固态薄膜的光致发光量子产率高达82%,辐射衰减速率常数高达105 s−1。通过变温发射光谱、时间分辨光致发光衰减和计算研究,发现这些金(III)树状大分子表现出热激活延迟荧光(TADF)性质。并基于这些金(III)树状大分子,制备出溶液加工型有机电致发光二极管(OLED),其最大电流效率为52.6cd A−1,最大外部量子效率为15.8%,高功率效率为41.3 lm W−1。并记录了这些OLED的运行稳定性,基于零代和第二代树状大分子的器件在100 cd m-2下的最大半衰期分别为1305小时、322小时。
为了更深入地了解这些包含三齿配体的金(III)树状大分子的电子结构,以及吸收和发射的起源,作者进行了密度泛函理论(DFT)和TDDFT 计算。对基态、激发态分子结构、激发态,以及参与激发的分子轨道进行了系统性的分析,其中480-520 nm 的激发主要由HOMO → LUMO贡献。HOMO是主要位于咔唑部分的π轨道,而LUMO是主要位于中央苯环和C^C^N配体的吡啶基部分的π*轨道,因此,HOMO → LUMO跃迁可以认定为 LLCT [π(咔唑) → π*(C^C^N)] 跃迁,三种结构的吸收带及其光谱分配与实验结果趋势一致。
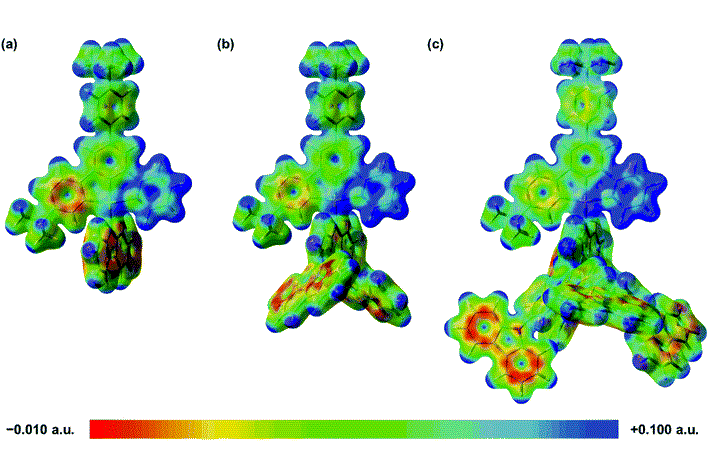
为了更深入地了解增加咔唑基单元对环金属化配体电子密度的影响,还计算了三种结构的基态静电势面。显然,由于树状咔唑取代基的吸电子作用,较高代咔唑基树枝的引入,导致更缺电子的吡啶基,这降低了辅助咔唑基N-供体配体上的电子密度,减少了其给电子的能力。咔唑基N-供体配体的供体强度较差,会使金属中心更加缺电子,通过σ效应从C^C^N钳形配体中吸取更多的电子密度。电子富集较少的金属中心对金属 dπ 轨道起到稳定作用,这反过来会导致 π* (C^C^N) 轨道较小程度的不稳定。总体而言,第二种结构的 HOMO-LUMO能隙 (3.04 eV)比第一种 (3.06 eV) 和第三种 (3.09 eV) 的能隙略窄,与实验趋势非常吻合。
为了更深入地了解发射态的性质,使用非限制性PBE0对T1进行了几何结构优化。三种分子的自旋密度主要位于 C^C^N 配体的咔唑基部分、中心苯环和吡啶基部分,从而支持发光态的LLCT [π(咔唑) → π*(C ^C^N)]特性。发射波长采用S0和T1优化几何结构之后的能量差近似,发射波长从第三种分子 (507 nm) 到第一种分子 (530 nm)与第二种分子 (534 nm)显现出红移特点,这与实验中观察到的趋势一致。
为了进一步了解 TADF 过程中涉及的激发态,使用包含Tamm-Dancoff 近似 (TDA) 的 TDDFT 优化了三种分子的S1和T1的几何结构。计算得到三种分子ΔE ST值分别为 0.005、0.006 和 0.004 eV。为了进一步了解第一种分子的的 ISC 和 RISC 过程的动力学,使用AMS软件ADF模块计算了ΔEST值、荧光速率常数 ( k fl ) 和磷光速率常数 ( k ph ) 、平均辐射衰减速率常数 ( k r,avg )、重组能 ( λ ) 以及kISC和kRISC。在 PBE0/TZP 水平计算的ΔE ST值为 0.006 eV,与实验值0.003 eV 非常吻合。计算出的kr,avg值4.3 × 104 s -1 的值也与实验确定的kr值8.0 × 104 s-1 在同一数量级上一致。计算出的kISC和k RISC 值分别为 4.4 × 109和 3.5 × 10 9 s-1,该高值与重金原子相关的大自旋轨道耦合常数具有一致性,并具有可比性。有效的 ISC 和 RISC 过程,促进该分子 中的 TADF发射。
- Lok-Kwan, Wing-Kei , Man-Chung Tang, Wai-Lung Cheung, Shiu-Lun Lai, Maggie Ng, Mei-Yee Chan and Vivian Wing-Wah Yam, Highly efficient carbazolylgold(III) dendrimers based on thermally activated delayed fluorescence and their application in solution-processed organic light-emitting devices, Chem. Sci., 2021, DOI: 10.1039/D1SC03690D
OLEDs双极性磷光基质材料的XPS和NEXAFS研究
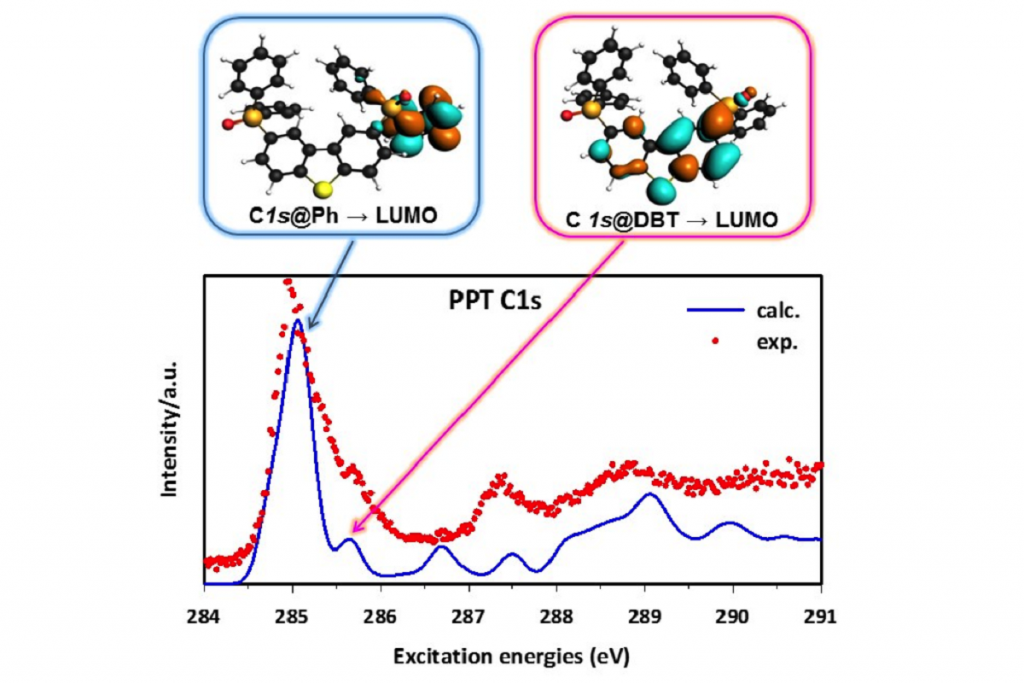
单通道的Kohn-Sham DFT理论结合跃迁势(Transition Potential),能够考虑核空穴形成的大部分的电子弛豫效应,从而描述轻原子的K-壳层NEXAFS光谱。这种方法提供了一组正交轨道,从中可以得到跃迁偶极矩。K壳层NEXAFS光谱是通过对每个非等效原子位的激发光谱进行单独计算,并将其贡献按相对权重相加得到的。这样就可以将总的光谱性质反卷积到不同组分中,从而有助于将光谱特征分解到分子的特定部位。
这种方法,最近被应用到2,8-bis-(diphenylphosphoryl)-dibenzo[b,d]thiophene(PPT)的C1s和O1s NEXAFS光谱的模拟,这是最近引入OLED中的一种双极性磷光主体材料,用于解释在Trieste的电子同步加速器气相束线处获得的实验光谱。PPT可以认为是由两个二苯基氧化膦(dPPO)部分,对小二苯并噻吩(DBT)核心官能化而形成。在C的K-边的DFT-TP计算表明,PPT的C1s谱主峰归属于dPPO臂的苯环,而第二弱峰则归属于PPT DBT核的苯环部分。
本研究的结论对OLED的未来应用具有重要意义:PPT的氧化膦基团是PPT的DBT核与外层基团之间π共轭的断裂点。然而,这些基团在很大程度上不影响DBT中心部分的电子性质。
- A. Guarnaccio, T. Zhang, C. Grazioli, F. Johansson, M. Coreno, M. de Simone, G. Fronzoni, D. Toffoli, E. Bernes, C. Puglia, PPT Isolated Molecule and Its Building Block Moieties Studied by C 1s and O 1s Gas Phase X-ray Photoelectron and Photoabsorption Spectroscopies, J. Phys. Chem. C 2020, 124, 9774−9786
石墨相氮化碳g-C3N4量子点依赖pH值的光致发光机理研究
石墨相氮化碳g-C3N4量子点是一种被广泛研究的荧光材料,其光致发光特性体现出pH值依赖性。然而不同的实验中,观察到性能相反的变化趋势,其机理尚不清楚。东南大学王金兰教授课题组基于含时密度泛函理论(TDDFT)和非绝热分子动力学模拟,提出中性和酸性条件下g-C3N4量子点光吸收与辐射/非辐射复合的协同机制。特别是在弱酸性条件下,g-C3N4量子点的强光吸收和弱非辐射复合,导致荧光发射较强。而在强酸性条件下,虽然光吸收仍然很高,但快速的非辐射电子空穴复合大大降低了激发态的布居,从而导致荧光猝灭。
N原子的质子化作用改变了跃迁通道的轨道组成和前线分子轨道重叠,从而调节了辐射和非辐射复合之间的竞争以及发光性能。此外,不同官能团的g-C3N4量子点的吸收和发射特性的变化趋势没有明显变化,这表明了该机理解释的普适性。DFT/TDDFT计算采用ADF,分子动力学模拟采用NAMD完成。
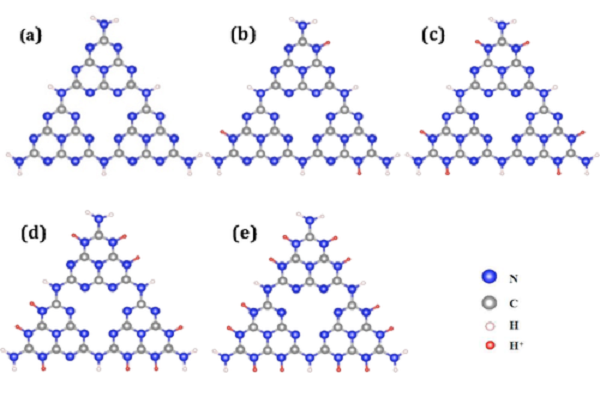
- Zhaobo Zhou, Xianghong Niu, Liang Ma, Jinlan Wang, Revealing the pH‐Dependent Photoluminescence Mechanism of Graphitic C3N4 Quantum Dots, Advanced Theory and Simulations, 2019, 2, 1900074