
概述
ADF 是世界上最早的 DFT 计算软件,计算功能非常完善、全面,建模、分析用户界面对初学者非常友好,节点内、跨节点并行计算效率非常高效。
- 一般功能:电子与结构、谱学与非线性光学性质、化学反应、热力学性质、溶剂化效应等,并包括高精度GW方法精确计算分子解离能与亲和势。
- 光学材料:零场劈裂、激发态辐射速率与寿命、系间窜跃、自旋轨道耦合,高精度GW方法精确计算ΔES-T。为OLED器件模拟软件Bumblebee提供原子层级模拟数据输入,辅助OLED器件模拟。
- 重元素与配合物:重元素体系研究必备工具,包含高精度相对论方法ZORA、X2C,完善的轨道成分分析、以及最流行的化学键分析方法EDA-NOCV、IQA等。
优势
- 效率优势:
- 支持节点内、跨节点高效并行计算,对较大体系,千核并行也能达到非常高加速比
- 支持大体系计算,例如大体系吸收光谱
- 普通工作站,甚至台式机就可以计算几百原子的规模的TDDFT性质
- 方法优势:
功能列表
- 分子轨道与能级分析
- 分子轨道MO投影到碎片轨道SFO、SFO可视化
- 基于片段轨道的CDFT:能够有效将电荷局限到体系的某个局部,并支持此类体系的分子轨道计算、能级计算、EDA 分析、ETS-NOCV 分析、转移积分计算等
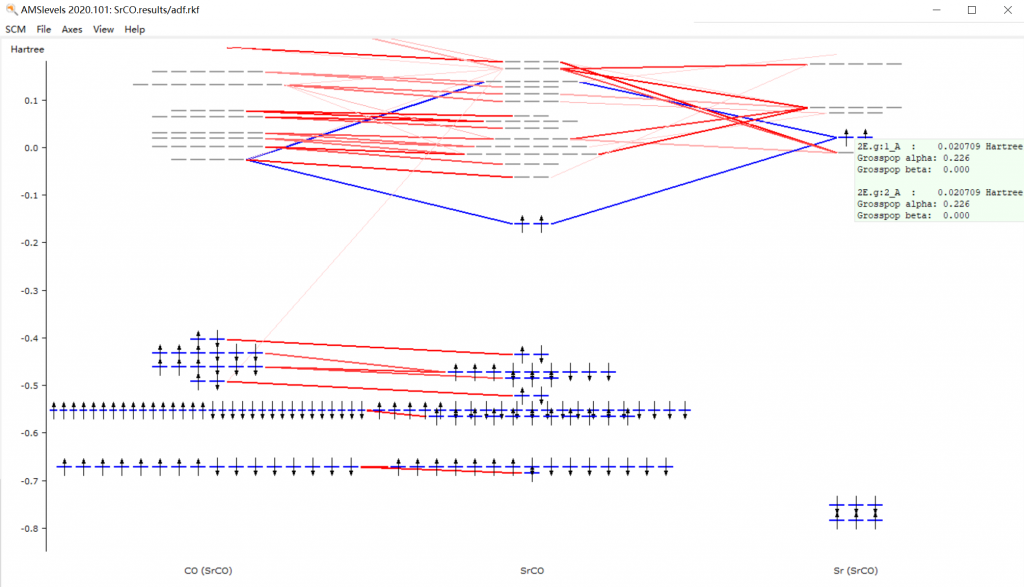
- 键合分析
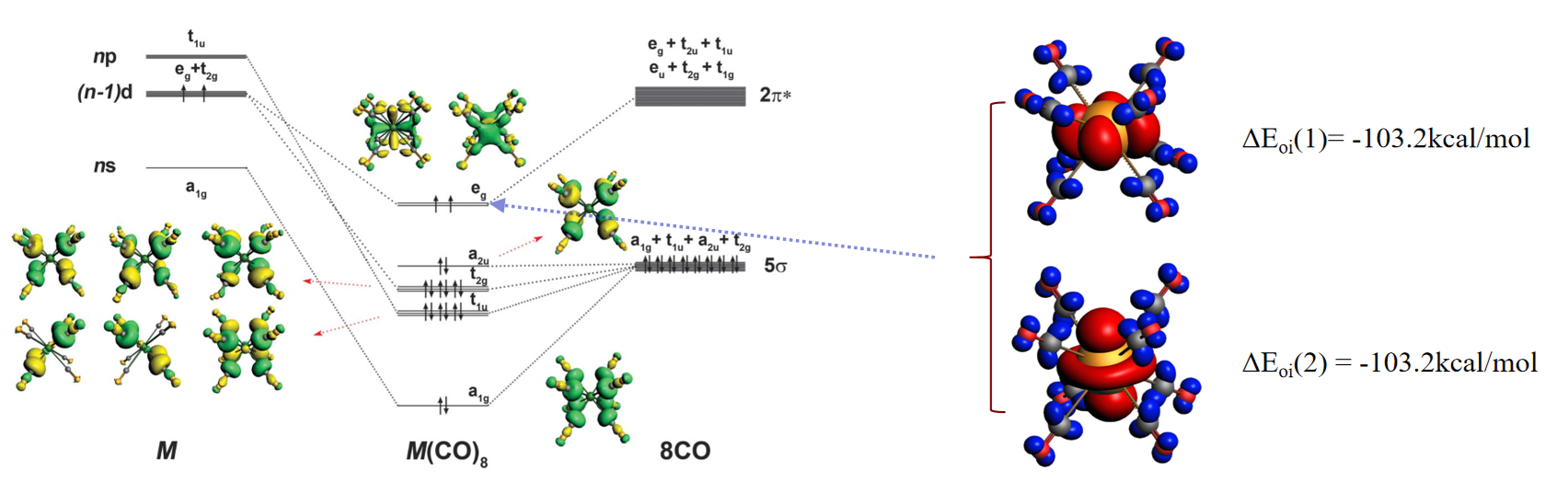
- 化学反应

- 激发态计算
- 紫外可见吸收谱(非相对论方法、相对论动能修正、考虑自旋轨道耦合)
- 自旋翻转跃迁
- ROKS-TDA 限制性开壳层的激发态计算
- X射线吸收(XANES、EXAFS、XPS)
- 激发态辐射跃迁寿命,荧光发射谱
- 解析梯度的TDDFT+TB优化激发态结构
- 开壳层大体系杂化泛函计算激发态加速近似HDA
- 考虑自旋轨道耦合的情况下计算ECD谱
- POLTDDFT方法快速计算Au、Ag团簇吸附小分子体系紫外可见吸收谱
- QMMM、多尺度方法计算紫外可见吸收谱、COSMO溶剂化的TDDFT
- 配体场DFT(LFDFT)对 d → d和f → d电子转移的情况,令计算结果更可靠
- 三重态零场劈裂ZFS
- OLED与系间窜越
- 磷光发射谱
- SOCME估算系间窜跃
- 激发态间跃迁偶极矩
- 系间窜越速率、Franck-Condon因子、Huang-Rhys因子
- 基于 ROSE 的激子转移积分
- GW-BSE 方法精确计算 S0-T1 能隙、精确 IP 值计算:顶点校正 GW 方法

- 谱学性质
- 非线性光学:
- 电荷、电子密度分析:
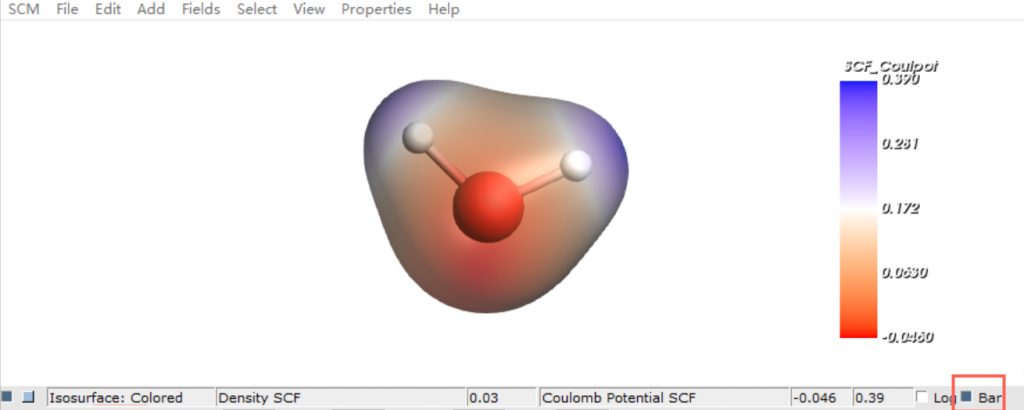
- 电荷迁移性:
- 转移积分方法计算分子间载流子迁移(教程)
- 金属-配体电荷转移(MLCT)、激发态电荷转移描述符
- 片段结合引起电子密度迁移:差分电子密度(教程)
- 溶剂化方法:
- 隐式溶剂化COSMO、SCRF、3D-RISM
- 显式溶剂化:QMMM、多尺度模拟、FDE、基于DRF与QM/FQ的QMMM
- 范德华色散系数
- 热力学性质:热容、Gibbs自由能、熵、焓等、溶剂化自由能
- 磁学性质:磁化率、Verdet常数、旋转g张量、Spinor:Spin Magnetization Density