
研究背景
氨燃料作为一种清洁能源具有广阔的应用前景,但其在实际应用中存在着燃烧速度慢、点火困难等问题。为提高氨的燃烧效率,研究人员提出通过加入二甲醚(DME)等反应性较强的组分来改善其燃烧特性。DME作为一种高质量的清洁能源,具有较高的热值和绝热燃烧温度,能够显著提高氨的反应活性,同时还能够有效减少NOx排放。这一研究的重点是通过先进的分子动力学模拟和密度泛函理论(DFT)方法,深入探讨DME/NH3混合燃料的燃烧机制,为未来绿色能源的开发提供理论依据。
研究方法
为揭示DME/NH3混合燃料在燃烧过程中的反应路径和化学机制,研究团队采用反应力场分子动力学(ReaxFF-MD)与密度泛函理论(DFT)相结合的数值模拟方法。ReaxFF-MD能够在原子尺度上模拟复杂的化学反应,为我们呈现分子之间的相互作用和化学键的演变过程。与此同时,DFT方法则用于计算分子反应的能垒、键解离能及反应位点。这种方法的结合不仅能够精确模拟燃烧过程中的微观机制,还能为实验结果提供有力的理论支持。
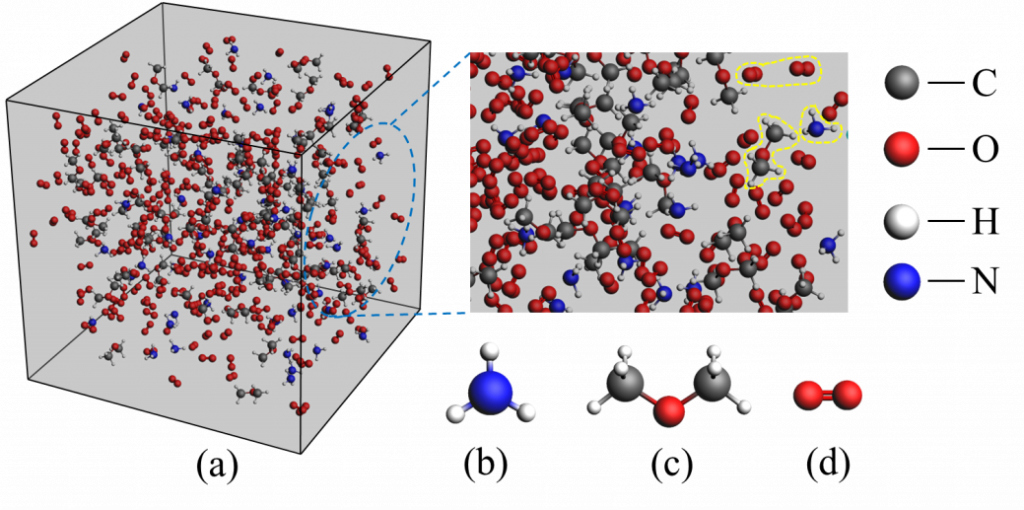
主要研究结论
通过对DME/NH3混合燃料的深入研究,研究团队为理解这种混合燃料的燃烧机制提供了新的视角:
① 温度对燃烧过程的影响:
研究发现,随着系统温度的增加,燃烧反应的速率显著加快,氧气的消耗量也随之增加。这表明温度的提升能够显著增强燃烧反应的活跃度。此外,在各种温度条件下,氨分子总是先于二甲醚分子被完全消耗。温度越高,二甲醚的消耗速度越快。
② 主要产物生成规律:
随着温度的升高,二氧化碳和水的生成量显著增加。氮元素在燃烧过程中主要形成氮气(N2)、一氧化氮(NO)和二氧化氮(NO2)等产物。随着温度的升高,氮气的生成量减少,而NO和NO2的生成量增加。这表明温度升高有助于NOx的生成。
③ 化学键的演变:
研究通过分析燃烧过程中化学键的变化,发现C-H、C-O和N-H键在反应过程中逐渐减少。随着温度的升高,C-H键完全消耗的时间显著缩短。N-H键的数量在反应过程中没有完全消失,部分以中间产物的形式存在。这表明在燃烧过程中,一些中间产物较为稳定,尤其是在较低温度下。
④ 初始反应路径分析:
二甲醚(DME)的初始反应路径主要以裂解反应为主,生成甲氧基(CH3O)和甲基(CH3)自由基。这一反应占据了所有初始反应中的49.06%,表明其在燃烧反应中占据主导地位。对于氨分子(NH3),初始反应路径则以氧化反应为主,生成H2O2和HO2等产物。这表明氨的燃烧主要通过氧化反应进行。
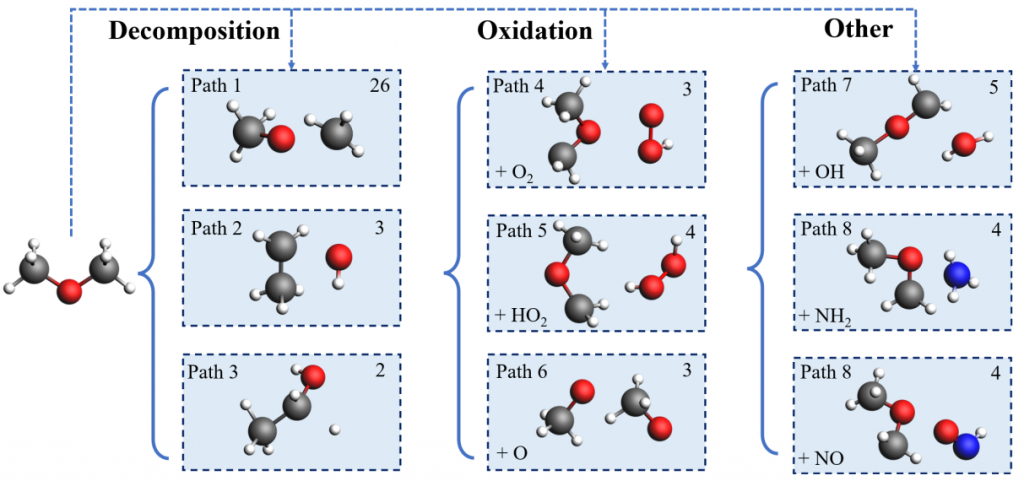
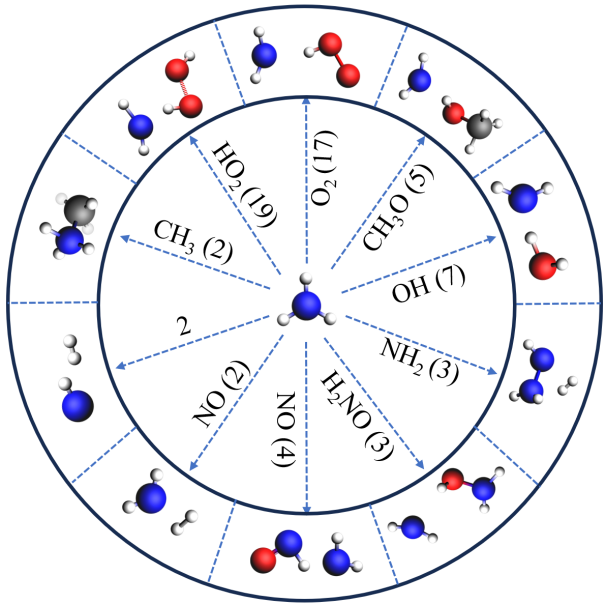
⑤ NOx排放的反应机制:
研究还发现,自由基(如羟基)在NOx的生成过程中扮演了重要角色。具体而言,羟基通过与HNO和HNO2分子的反应生成NO和NO2。这一反应途径在所有NOx相关反应中占据主导地位,尤其是NO的生成反应发生频率高达183次。
参考文献
Atomic insights into the combustion mechanism of DME/NH3 mixtures: A combined ReaxFF-MD and DFT study, International Journal of Hydrogen Energy, Volume 80, 28 August 2024, Pages 743-753
感谢重庆大学李海涛教授供稿,ReaxFF 分子动力学模拟采用 AMS 软件完成。