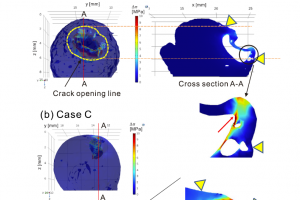
一、概述 岩浆囊的快速减压是脆性碎裂的重要机制,从而导致爆炸性火山喷发。由于岩浆是一种粘弹性流体,它是否符合固体的碎裂标准应取决于该过程的速率和时间。利用粘弹性流体进行的减压实验表明,碎裂和膨胀行为由三个特征时间的组合控制:减压时间 tdec、材料的弛豫时间 τr 和气泡的粘性膨胀时间 τv。类似脆性碎裂的发生是试样表面裂纹处有相当数量的气体突然释放引起,气泡分布的形态比较复杂,裂纹是由于连续相的韧性断裂或气泡的连接在试样内部萌生。 tdec < τr 发生类似固态的脆性碎裂 tdec > τv 气泡只发生膨胀而不碎裂 τr < tdec < τv 发生类似固态的碎裂,但有明显的时间延迟 在火山喷发建模中,考虑脆性碎裂尤为重要。而在阐明脆性碎裂机制时,将破裂起始点与试样的内部结构一一对应起来也至关重要。本研究通过 X 射线显微断层扫描(XCT)数据表征减压前试样的三维气泡结构,并使用高速摄影结合可见光照明记录快速减压过程中试样的动态响应,建立表面裂纹开口和内部气泡分布之间的联系。利用有限元法(FEM)数值模拟计算试样在减压过程中的应力演化和变形场,采用线性 Maxwell 模型描述试样的粘弹性能,讨论尺寸和空间分布对裂纹发展和类脆性碎裂触发的影响。 二、实验和模拟 2.1 试样 含有气泡的糖浆具有与岩浆接近的较大刚度 G 且粘度 η 范围很广,是一种适合模拟岩浆囊碎裂的材料。本研究所用试样为麦芽糖浆,将过氧化氢溶液(30%)作为添加剂混合以产生气泡,添加二氧化锰粉末作为过氧化氢溶液分解的催化剂。 2.2 快速减压试验 利用快速减压装置模拟碎裂,将试样放在容器的铝质底板上,容器顶部用塑料薄膜密封。使用精密压力控制器进行氮气加压,通过焦耳加热粘附在薄膜上的细镍铬丝使薄膜破裂。由于薄膜的突然破裂,容器中的加压气体快速释放,从而对试样进行快速减压。 减压过程中装置内部的压力曲线可以用指数衰减曲线相当准确地近似: 其中,p0 为减压前的初始压力,pa 为环境大气压,tdec 为特征减压时间。p0 设定为 2.1 MPa,特征减压时间 tdec 约为 3 ms。 使用 SPring-8 的 BL20B2 光束线进行 XCT 扫描,像素尺寸为 15.5 μm。采用 12 位数字高速摄像机观察试样在减压过程中的动态行为,空间分辨率为 0.10 […]