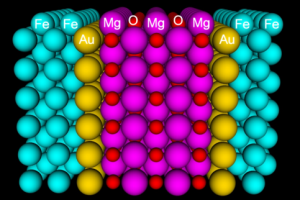
概述 块体材料的研究已经非常成熟,因此材料的表面与界面的重要性就格外的凸显出来。新型材料越来越复杂,不同材料组成的异质界面成为许多功能器件的基础,最常见的一个例子是半导体领域里的含有高k介电材料的多层门电极堆叠。由于界面层往往很薄,其中的缺陷起着十分重要的作用,这需要在原子级别上描述结构,才能充分考虑杂质、空位等对性质的影响;同时,只有进行基于量子力学的计算模拟,才能计算例如肖特基势垒和漏电流等。在原子级别上对材料表面和界面进行量子力学的模拟正是 QuantumATK 最为擅长的领域。 片层(Slab)模型 与其他的周期性模型程序类似,QuantumATK 也可以用传统的 Slab 模型来描述界面体系,但Slab模型有很大的缺陷和局限: Slab 最大的不足是无法模拟实际表面下方通常是无限大的块体材料; 由于厚度有限,Slab 中的电子容易体现出量子限制的效应; 两个表面之间、表面与界面之间存在相互影响; 很难正确的在表面方向模拟外加电场; 经常需要表面钝化、偶极校正等额外补救措施。 双电极界面(Two-probe interface)模型与性质计算 采用双电极界面模型模拟材料界面,比Slab模型更加便捷,可以避免Slab模型的上述问题。此外,双电极模型还可以更好的研究: 异质结的电流-电压特性,例如: 漏电流 金属-半导体界面的肖特基势垒 磁性隧道结的自旋输运 缺陷(杂质和空位)对输运性质的影响; 界面上的电荷转移 使用QuantumATK研究界面体系的优势 通用、高效的计算引擎 QuantumATK 计算基于第一性原理,因此可以用于研究全新材料的各种性质,例如: 传统金属-半导体界面 高k介电材料 金属、半导体纳米线 纳米管、金属纳米管接触 原子簇 等等 QuantumATK 中使用局域基组展开方法,尤其适用于研究局域化缺陷(杂质、空位等),ATK-DFT计算引擎可以计算千原子级别的体系的性质。ATK-SemiEmpirical 则可以计算更大的体系。 NanoLab高级图形用户界面:专注于研究,更快获得结果 NanoLab 图形用户界面丰富易用的功能可以让用户专注于研究项目的科学问题,专心思考科学问题,更快的发现新材料、创建新结构,避免在数据的导入、导出、处理、作图等琐碎的问题上浪费时间。NanoLab 可以: 方便快捷的材料表面建模工具 强大的材料界面结构建模工具 最合理的界面结构优化方法 快速构建各种结构模型 内嵌晶体结构数据库 搜索在线晶体结构数据 亮点文章 QuantumATK 在材料界面的最优超胞建模中的应用(D. Stradi, L. Jelver, S. Smidstrup and K. […]