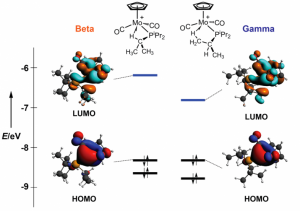
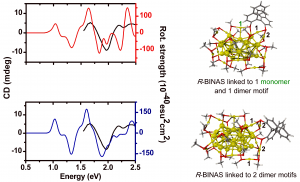
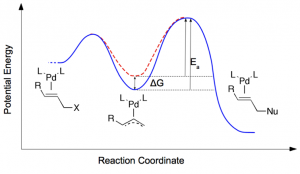
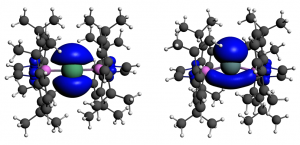
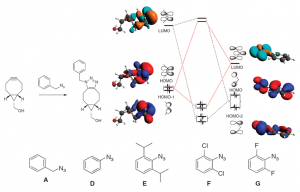
参考文献: E. F. van der Eide, P. Yang, and R. M. Bullock, Isolation of Two Agostic Isomers of an Organometallic Cation: Different Structures and Colors,Angew. Chem. Int. Ed. 52, 10190-10194 (2013) 本文第一次分离、表征出两个元结构异构体(agostomers)。Mo阳离子钢琴凳化合物异构体是一种加氢催化的重要中间物,通过结晶析分离,并能通过溶解互相转化。[CpMo(CO)2(PiPr3)]+ h的γ-元结异构体颜色是蓝色,β-元结异构体是橙色。 ADF计算确认这两种元结异构体能量非常接近,计算得到的VIS/NIR谱与实验数据非常一致。两种异构体的前线轨道也非常相似,β-元结异构体的LUMO比γ-元结异构体能量高0.64eV。这导致吸收峰能量更高。 β-元结异构体吸收可见光中较短波长,导致β-元结异构体的颜色是橙色,γ-元结异构体为蓝色。 使用ADF功能:ZORA, TDDFT, VIS/NIR, chemical bonding analysis