
参考文献:
D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Isolation and characterisation of a uranium(VI)-nitride triple bond, Nature Chem. 5, 482-488 (2013)
D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake, and S. T. Liddle, Synthesis of a Uranium(VI)-Carbene: Reductive Formation of Uranyl(V)-Methanides, Oxidative Preparation of a [R2C═U═O]2+ Analogue of the [O═U═O]2+ Uranyl Ion (R = Ph2PNSiMe3), and Comparison of the Nature of UIV═C, UV═C, and UVI═C Double Bonds,J. Am. Chem. Soc. 134, 10047−10054 (2012)
O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake, and S. T. Liddle,Uranium-Carbon Multiple Bonding: Facile Access to the Pentavalent Uranium Carbene [U{C(PPh2NSiMe3)2}(Cl)2(I)] and Comparison of UV=C and UIV=C Double Bond, Angew. Chem. Int. Ed. 50, 2383-2386 (2011)
D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake, and S. T. Liddle, A delocalised arene-bridged diuranium single molecule magnet, Nature Chem. 3, 454-460 (2011)
Relativistic DFT, bonding analysis: orbital levels, bond orders, charges, QT-AIM, NBO
D. Patel, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Reductive assembly of cyclobutadienyl and diphosphacyclobutadienyl rings at uranium, Nature Commun. 4, 2323 (2013)
D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Isolation and characterisation of a uranium(VI)-nitride triple bond, Nature Chem. 5, 482-488 (2013)
Prof. Steve Liddle及其合作者在一系列突破性文章中报道了新的铀化合物的合成、分离及表征。
在一系列的坚持不懈的证实、理解f区元素的奇特化学成键的努力中,首次发现了fcyclobutadienyl化合物(Nat. Commun. 4, 2323),末端U-N三键化合物(Science 337, 717, Nat. Chem. 5, 482),O=U(VI)=C(PR2)2 (J. Am. Chem. Soc. 134, 10047),U(V) carbenes (Angew. Chem. Int. Ed. 50, 2383)。
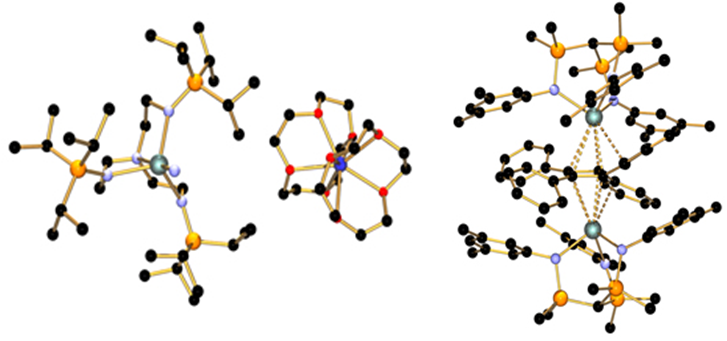
左:第一个U(V)-N三键;之后氧化成第一个U(VI)-N三键;右:第一个U-cyclobutadienyl化合物
ADF的相对论DFT计算为这些新铀化合物的化学键,以及与以前发现的那些铀键、同类相似的d区化学键提供了有价值的视角。成键相互作用分别使用键级(Mayer、Nalewajski-Mrozek)、QTAIM、NBO、orbital populations、MDC charges(ADF的Single Point计算默认给出)以及spin density等方式进行研究。
铀化合物的成键相互作用:
虽然5f电子主要影响非键,但却是参与到U-X多键以及芳香环π电子配体。
铀的氧化态能够强烈地影响键级,以及相关的5f、6d轨道参与π键。U-X距离很短的时候,N的2Pz轨道与5f和6d轨道有反键相互作用,把σ键的能量推的比π键高,从而形成U-N三键。虽然U-N距离在U(V)氧化到U(VI)时,从1.825缩短到1。799埃,但Mayer键级几乎不受影响(2.91,2.92)。
在(Nature Chem. 3, 454)中的反三明治结构化合物中,U的5f轨道与中心芳环有强共价、δ-backbonding作用,两个U中心三明治cyclobutadienyl之间的相互作用,由于尺寸不匹配、轨道重叠小,所以主要是静电相互作用。
![从实验分子结构[U(N)(TrenTIPS)]; TrenTIPS = {N(CH2CH2NSiiPr3)3}3−, iPr = CH(CH3)2))截取的U(VI)-N末端模型化合物三键相互作用;σ键(HOMO,最右上)能量高于两个准简并π键(左下)](http://www.fermitech.com.cn/wp-content/uploads/2016/08/U_N_bond.png)
从实验分子结构[U(N)(TrenTIPS)]; TrenTIPS = {N(CH2CH2NSiiPr3)3}3−, iPr = CH(CH3)2))截取的U(VI)-N末端模型化合物三键相互作用;σ键(HOMO,最右上)能量高于两个准简并π键(左下)
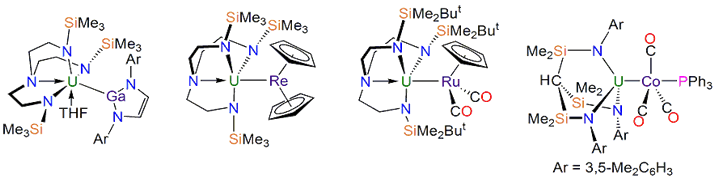
Liddle课题组发现的更有趣的U键;从左到右:U-Ga、U-Re、U-Ru、U-Co
关键词:成键分析,重元素,相对论DFT