

ADF软件高级培训班(2016年南京站)邀请通知
ADF软件作为计算化学和计算材料学模拟平台以其专业性能得到各大高校及科研院所老师和同学们的青睐。费米科技定于2016年9月22-23日在南京举办为期两天的ADF软件高级培训班,以帮助更多的科研人员全面的了解ADF软件在计算化学各领域的应用。 主讲人介绍: 刘俊,ADF软件中国区技术支持。毕业于北京大学化学与分子工程学院。熟悉计算化学理论以及密度泛函理论的程序化。长期担任ADF软件技术支持,2010年曾到荷兰ADF软件开发组学习。 培训内容: ADF参数物理意义,以及如何设置 ADF生成各种图像数据及其处理 片段相互作用与健能分解 氢键的电子转移NOCV分析 转移积分与电子迁移率 过渡态搜索 荧光与磷光寿命、辐射跃迁速率 自旋轨道耦合的应用(SOCME) 开壳层的NRM计算 激发态几何结构优化 势能面交差(激发态反应) 溶剂化 溶液中的Gibbs自由能 表面吸附结构的健能分解、NOCV 反应力场案例 培训时间:2016年9月22-23日 上午 9:00-12:00 下午13:30-17:00 培训地点:南京工业大学 虹桥校区 教Ⅲ-306室 培训费用:免费(食宿交通等费用自理) 人数限制:50人 报到时间:9月22日 08:00-08:50 温馨提示: 为了方便广大用户对软件更好的学习与后续使用,请自带笔记版电脑,现场安装最新版ADF试用license。 后续培训信息: ADF软件高级培训班(2016年成都站)时间待定 ReaxFF模块上机培训 费米科技作为第四届国际分子模拟大会参展商将参加“反应力场培训研讨会——方法、应用与挑战(https://www.icms2016.org/workshop.php?lang=zh)”并进行上机培训。 本次培训已经圆满结束!
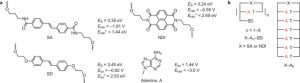
ADF Highlight:DNA发夹结构中深能级空穴转移导致超快电荷迁移(Nature Chemistry,2016)
参考文件: Nicolas Renaud, Michelle A. Harris, Arunoday P. N. Singh, Yuri A. Berlin, Mark A. Ratner, Michael R. Wasielewski, Frederick D. Lewis & Ferdinand C. Grozema, Deep-hole transfer leads to ultrafast charge migration in DNA hairpins. Nature Chemistry(2016)doi:10.1038/nchem.2590 通过DNA双螺旋的电荷转移在化学与生物化学中是非常基本的研究课题,而且也有潜在的应用,例如DNA纳米电子学。在DNA纳米电子学领域,人们对如何影响、增强电荷转移有相当的兴趣。本文中,展示了DNA电荷转移的一种新机制,也就是所谓的“深能级空穴转移”,空穴通过核碱基的能量很低的电子态,发生长程转移。 本文结合实验与理论计算研究表明,通过改变电荷入射的能量相关性质,可以实现这样的电荷转移。这种方式可以将转移率提高两个数量级,并且对距离的依赖很弱。这种转移比以前的转移机制(弛豫到低能量态的方式)要快很多。这开启了优化DNA电荷转移的新方向,对电荷转移率的提高达到了史无前例的水平。 本文使用了ADF模块 的转移积分功能。 关键词:DNA nanostructures,Electron transfer
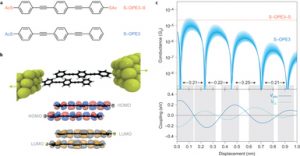
ADF Highlight:机械控制π堆叠二聚体的量子干涉(Nature Chemistry,2016)
参考文献: Riccardo Frisenda, Vera A. E. C. Janssen, Ferdinand C. Grozema, Herre S. J. van der Zant & Nicolas Renaud.Mechanically controlled quantum interference in individual π-stacked dimers.Nature Chemistry. Nature Chemistry(2016) doi:10.1038/nchem.2588 近期观察到单分子结的破坏性量子干涉,肯定了量子效应在分子体系的电子电导率中的角色。这些效应是很多化学与生物学过程的核心,可能对设计单分子电子元器件用于分子尺度的内秉量子效应的研究也有帮助。 本文展示了量子干涉在同一个分子体系内,可以通过机械地调整结构来打开或关闭。结合“从头算”与单分子电导测量,沿着二聚体π堆叠打破的路径,破坏性量子干涉展现出准周期性。这样的结果表明,通过研究二聚体π堆叠的微结构-性质关系,可以在埃以下的解析度,超过一个数量级的强度上控制分子电导率。 本文使用了ADF模块 的转移积分功能。 关键词:Electron transfer,Electronic devices,Molecular electronics
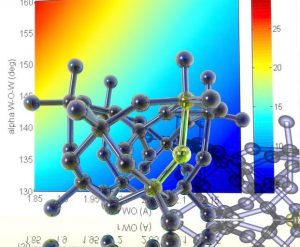
ADF Highlight:钨多金属氧酸盐的NMR的spin-spin耦合(Angew. Chem. Int. Ed.,2005)
参考文献: A. Bagno and M. Bonchio, Vicinal Tungsten.Tungsten Coupling Constants in Polyoxotungstates: DFT Calculations Challenge an Empirical Rule.Angewandte Chemie International Edition 44 (13), 2023 (2005). 多金属氧酸盐(Polyoxometalate,POM)是基于MO6八面体的团簇族,其中M是钨或钼,中心通常包括一个杂环原子。POM在材料化学、催化和药物学中有广泛应用。这些化合物的分子结构通过W-183细胞核的NMR谱探测得到,。但实际上本文也通过计算化学现在已经能够高精度地预测像gamma-[SiW10O36]8-这么大的分子。 意大利Padova大学的Prof. Bagno评论:“Through the unique capability of ADF (and associated CPL module) to compute the electronic structure of molecules containing heavy atoms in a relativistic DFT framework we have been […]
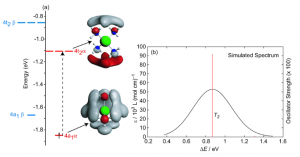
ADF Highlight:双核金属自由基成键(Angew. Chem. Int. Ed.,2012)
参考文献: E. F. van der Eide, P. Yang, E. D. Walter, T. Liu, and R. M. Bullock, Dinuclear Metalloradicals Featuring Unsupported Metal-Metal Bonds Angew. Chem. Int. Ed. 51, 8361-8364 (2012) 金属-金属相互作用在顺磁多核过渡金属配合物中,对金属蛋白分子的反应性非常关键。对这种作用的理解,对含金属功能聚合物的研发非常重要。尤其是这种不桥联、通过金属-金属键连接的顺磁二聚体的成键和谱学性质还没有被很好的理解。 本文首次合成不能给分离出稳定的不桥联的双金属自由基[{CpW(CO)2(PMe3)}2]•+ (1•+)。相对论密度泛函计算表明,SOMO是M-M反π轨道,并自动得到光谱性质。TDDFT计算得到的光谱与实验一致,并指认出λmax = 966 nm的近红外吸收峰是π → π* 跃迁到SOMO贡献出来的。自旋-轨道耦合DFT计算得到的EPR参数也与实验一致。 上图是测量与计算(垂线)得到的[{CpW(CO)2(PMe3)}2] (1) ,以及稳定的金属-金属氧化物形成的自由基[{CpW(CO)2(PMe3)}2]•+ (1•+)的VIS/NIR谱。966nm NIR特征跃迁是π → π*跃迁到SOMO 使用ADF的功能:spin-orbit coupling, TDDFT, EPR, molecular orbital analysis
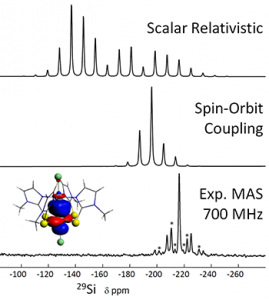
ADF Highlight:Pt的σ-donation的29Si NMR包括强相对论效应(Angew. Chem. Int. Ed.,2011)
参考文献: L. A. Truflandier, E. Brendler, J. Wagler, J. Autschbach, 29Si DFT/NMR Observation of Spin-Orbit Effect in Metallasilatrane Sheds Some Light on the Strength of the Metal→Silicon Interaction Angew. Chem. Int. Ed. 50, 255 (2011) 相对论效应通常而言随着原子序数的平方增大,因此对于重元素的化学结构和反应性有实质性的影响。但轻元素与重元素相互作用的时候,相对论效应会传递到轻元素上,也就是所谓的“the heavy-atom effect on light atoms (HALA)” 实验工作结合理论的计算,本文制备、分析了三个σ-donating键过渡金属配合物:TM→Si(TM=Ni、Pd、Pt)。健能分解、NBO分析确认了从Ni、Pd、Pt到Si的配位——虽然Pt配合物也存在明显的共价作用。 三种配合物,29Si NMR信号都有高场偏移,比Si高价模型要多得多。ADF的ZORA相对论DFT计算揭示:Scalar相对论计算就足够重复出观察到的NMR性质,也就是不考虑自旋- 轨道耦合,当然考虑自旋轨道耦合更精确,但计算量更大(约4倍)。因此,可以推知,这个体系中,自旋-轨道耦合对NMR的影响可以忽略。但考虑自旋-轨道耦合(使用ADF中的ZORA Spin-Orbit方法),29Si的墙屏蔽就能观察到了,(尤其是对Pt的情况)实验与计算结果就符合的更好了。 计算得到的1JPt,Si耦合常数也与实验一致。 使用ADF的功能:spin-orbit coupling, NMR, bonding analysis, NBO
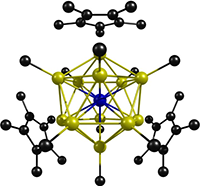
ADF Highlight:十二个单电子配体的钼配合物(Angew. Chem. Int. Ed.,2008)
参考文献: T. Cadenbach, T. Bollermann, C. Gemel, I. Fernandez, M. von Hopffgarten, G. Frenking and R. A. Fischer, Twelve One-Electron Ligands Coordinating One Metal Center: Structure and Bonding of [Mo(ZnCH3)9(ZnCp*)3].Angewandte Chemie International Edition, 47 (47), 9150 (2008). 本文报道了一个独特的钼-锌配合物,锌化合物通过锌原子与中心钼原子相连,通过x射线描述了这种12配位结构。 多达九个的单原子螯合配体已经有100多年的历史了,并且作为金属-金属团簇也非常著名(一种金属原子被另一种金属原子形成的壳包围,也被后者的电子包围)。这个分子另一个独特之处是它的电子结构。基于部分计算结果,中心原子与配体之间的平均键级为1/2,钼中心的sd5杂化轨道参与到和周围锌原子形成的共价键。另外,周围的锌原子形成了6电子二十面体的电子结构,导致他们之间也形成了弱键。 如此,这个钼锌配合物与传统的金属-配体配合物性质类似-金属与配体之间主要都是共价键;同时又与富勒烯金属-金属团簇相似-周围金属原子之间存在键。Prof. Frenking说:“the ADF calculations have been crucial for understanding the bonding situation in the molecules […]
ADF Highlight:锂-氨溶液在分子水平的模型(Angew. Chem. Int. Ed.,2009)
参考文献: E. Zurek, P. P. Edwards and R. Hoffmann, A Molecular Perspective on Lithium-Ammonia Solutions. Angewandte Chemie International Edition, 48(44), 8198 (2009). 美国康奈尔大学Roald Hoffmann教授是诺贝尔化学奖得主,是ADF的忠实用户。本文介绍了关于锂-氨溶液的详细计算分析工作。锂金属-氨溶液从200年前首次发现的时候,就一直被人们好奇于它的“fine blue colour”。 最终结论是:这些体系不是“金属铵”,而是碱阳离子以及溶液中电离的电子,这增加了这个体系的神秘性。电子态的怪异性、卓越的颜色性质,都能够使用TDDFT进行研究。 锂离子的氨溶液结构优化(使用COSMO溶剂化模型),以及电子吸收光谱的计算,都使用ADF完成。其中一些模拟中,涉及到自由基体系,溶剂化电子分布在氨分子周围。开壳层电子吸收谱的计算也还不算普遍。光谱和涉及的电子转移的轨道的可视化是在ADF-GUI中完成的。 使用ADF功能: excited states (with TDDFT), un-paired electrons (open-shelled systems)
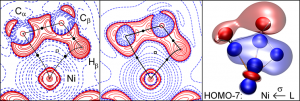
ADF Highlight:电荷密度和NMR解释Ni-元结键(Angew. Chem. Int. Ed.,2011)
参考文献: W. Scherer, V. Herz, A. Bruck, C. Hauf, F. Reiner, S. Altmannshofer, D. Leusser, and D. Stalke, The Nature of β-Agostic Bonding in Late-Transition-Metal Alkyl Complexes Angew. Chem. Int. Ed. 50, 285 (2011) 为了确认β-元结键的性质,Scherer和同事合成了[(DCpH)Ni(dtbpe)]+[BF4]– (DCp = dicyclopentadiene, dtbpe =tBu2PCH2CH2PtBu2),一种硬键β-元结化合物。结合实验和理论,对电子密度和NMR位移进行了研究。 为了更好地符合实验的结果,NMR计算有必要使用ADF中的hybrid GGA,并考虑自旋-轨道耦合。反铁磁 (σd),顺磁 (σp),spin-orbit (σSOC) 对前过渡金属和后过渡金属化合物屏蔽张量的贡献揭示出:元结H的1H NMR化学位移主要由σp 决定,H的原子电荷和σd 对各种模型体系几乎保持为常数。‘hydridic shift’项解释高场偏移因此就是不正确的了。实验的电荷密度分析与ADF的AIM计算结果完美地相符,有助于进一步将β-元结相互作用归类。 对于后过渡金属化合物,在M-H键区域显著地产生电子密度,消耗了一个C-H键。分子轨道(MO)分析揭示出:对于金属 -烯烃体系,后过渡金属β-元结键使用经典的Dewar-Chatt-Duncanson模型能够被描述的很好。争议是M←L σ-donation项。 使用ADF的功能:AIM, spin-orbit coupling NMR, bonding analysis