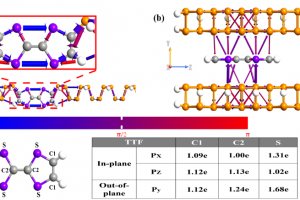
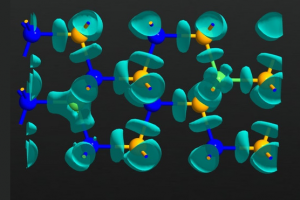
范德华${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$异质结构中磁相互作用的调节:从头算方法的比较研究 作者研究了机械应变、堆叠顺序和外部电场对二维(2D)范德华异质结构的磁相互作用的影响,其中二维铁磁性金属 ${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$ 单层沉积在锗烯上。使用了三种基于从头算方法的不同计算方法,(i)格林函数方法,(ii)广义布洛赫定理,和(iii)超胞方法,并进行了仔细的比较。首先,计算了独立${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$单层晶胞内三个Fe原子的壳层分辨交换常数。作者发现,用方法(i)和(ii)获得的结果在质量上是一致的,并且与以前报道的值在质量上也是一致的。垂直于${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$/锗烯异质结构施加 E=±0.5V/Å 的电场会导致交换常数的显著变化。结果表明,${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$/锗烯中的Dzyaloshinskii-Moriya相互作用(DMI)主要由最近邻主导,导致方法(i)和(ii)之间的定量一致性良好。此外,作者证明了DMI可通过应变、堆叠和电场调节,从而产生铁磁/重金属界面相当的大DMI,铁磁/重金属界面已被公认为承载孤立skyrmions的典型多层体系。几何变化和杂化效应解释了DMI在界面处的高可调性的起源。通过方法(iii)获得的电场驱动DMI与方法(ii)中使用的更精确的从头算方法在质量上一致。然而,方法(iii)高估了DMI的场效应约50%。这种差异归因于方法(ii)和(iii)中应用的从头计算方法中使用的电场和基组的不同实现方法。磁各向异性能(MAE)也可以通过在${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$/锗烯异质结构中施加压缩或拉伸应变而显著改变。相反,对于高达 1V/Å 的电场,施加电场只会导致MAE发生相对较小的变化。 Li, D.; Haldar, S.; Drevelow, T.; Heinze, S. Tuning the Magnetic Interactions in van Der Waals ${\mathrm{Fe}}_{3}{\mathrm{GeTe}}_{2}$ Heterostructures: A Comparative Study of Ab Initio Methods. Phys. Rev. B 2023, 107 (10), 104428. https://doi.org/10.1103/PhysRevB.107.104428. 通过相邻掺杂策略引入六方氮化硼的可调谐d0磁性 为了满足由于不断进步的自旋电子学对稀磁半导体(DMS)的需求,设计具有高稳定性、自旋极化和居里温度的d0-DMS是至关重要的。目前引入d0磁性的研究仅限于单原子掺杂,缺乏对局部磁矩和长距离磁耦合的调控措施。在此,采用相邻掺杂策略来引入用于调节d0-DMS的磁性能的自由度。作者观察到,通过分别引入Si和O原子作为中心和相邻掺杂剂,本征非磁性六方氮化硼(h-BN)表现出显著的局部磁矩。此外,还观察到掺杂h-BN的电离能、总磁矩、磁耦合和居里温度对Si–O配位敏感。随后,通过高Si–O配位掺杂设计了具有高热稳定性、100%自旋极化、长程铁磁耦合和高居里温度的磁性半金属(Si–O3掺杂的h-BN)。本研究以Si–O相邻掺杂h-BN的设计为例,提出了一种引入可调谐d0磁性的可行方法。 Wang, B.; Ning, J.; Zhang, J.; Wang, D.; Zhang, C.; Hao, Y. Tunable […]