
概述
锂离子电池因其高容量、高功率的优点,在储能领域发挥着至关重要的作用,广泛用于电动汽车和便携式电子产品等。电动汽车的快速发展要求高能量密度、高循环寿命和低成本,进一步改进制造工艺和电极设计仍然存在挑战。
在电极制造过程中,压延工艺是定制电极微观结构的关键步骤。在本研究中,使用离散元法(DEM)和粘结颗粒模型研究压延过程对电极微观结构的影响,对使用 X 射线计算机断层扫描(XCT)表征的真实电极结构和理想化 DEM 结构进行综合评估,计算分析基于断层扫描和 DEM 的电极结构及输运特性,即孔隙率分布、比表面积和曲折因子。在考虑碳粘合剂域(CBD)相之后,进一步分析电化学性质。
亮点
- 对基于高分辨率 XCT 表征和 DEM 的锂离子电池阴极结构进行评估
- 在 Simpleware 软件中生成高质量的四面体网格,将网格模型导入 COMSOL 中进行仿真
- 考虑不同的压延水平和 CBD 相,分析电化学性能
方法
图像处理
电极结构由 96 wt% LiNi0.6Mn0.2Co0.2O2(NMC622,BASF)、2 wt% C65 炭黑(Imerys)和 2 wt% PVDF(Solvay)配制,使用带有 MTI MSK-HRPMR100DC 压延设备的辊压机将干燥的电极压延两次。通过 XCT 对阴极结构进行表征,采用未压延结构和压延结构与 DEM 预测进行比较。使用基于机器学习的图像分析工具 Ilastik 预测扫描图像中不同相的体积分数,即 AM 颗粒相、CBD 相和宏孔相。
原始扫描图像(图a)经过过滤和二值化后获得 AM 颗粒相(图b),进行分割处理分离和标记单个 AM 颗粒(图c),并获得其各自的体积和坐标。在 DEM 模拟中,阴极结构内的颗粒近似为球形颗粒(图d)。

图1:AM 颗粒相的图像处理步骤
DEM 模拟
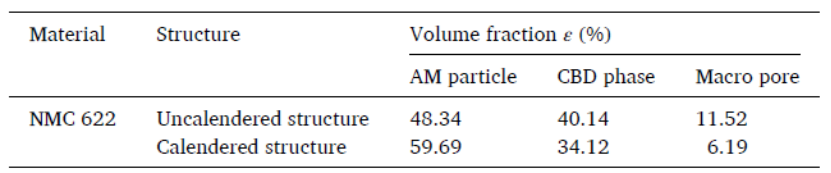
采用键模型描述 CBD 相间的相互作用(图a),Fb 和 Mb 是键合力和键合扭矩。在 Altair EDEM 软件中导入断层扫描处理的未压延电极结构,将颗粒间的键分配给电极结构(图b)。将根据等效球形颗粒计算得出的粒度分布 (PSD) 与使用激光衍射实验表征的 PSD 进行比较,表现出良好的一致性(图c)。通过 DEM 以 0.01 m/s 的恒定速度向下移动顶板模拟压延过程,得到孔隙率-压力关系(图d),与实验结果的对比表明该 DEM 模型可以捕捉压延过程中孔隙率的演变。
微观结构分析
使用 MATLAB 应用程序 TauFactor 基于 3D 体素化的图像数据计算比表面积和曲折因子,分析压延前后的电极微观结构。
电化学表征和模拟
为制造纽扣电池,将电极切成 15 mm 的圆盘,Celgard 分离器切成 19 mm 的圆盘以避免短路。电池组装完成后,阴极半电池在 2.5 至 4.2 V 的电压窗口(针对Li/Li+)内,以 C/20(C/50 截断)的 C 速率进行两次 CC-CV 充放电循环,其中半电池包含锂金属对电极。使用 BCS-805 Biologic 电池循环仪进行电化学测试,然后以恒定电流 C/10 和对应于 C/10、C/5、C/3 和 1C 的放电 C 速率对电池进行充电。
使用 Simpleware 软件对分割的体积图像进行网格划分,生成大约 1000 万个线性四面体单元,包含 520 万个自由度。将网格模型导入 COMSOL Multiphysics 软件进行基于扫描图像的有限元模拟。采用 Pardiso 求解器求解离散化输运和电极动力学方程。
结果
AM 颗粒结构分析
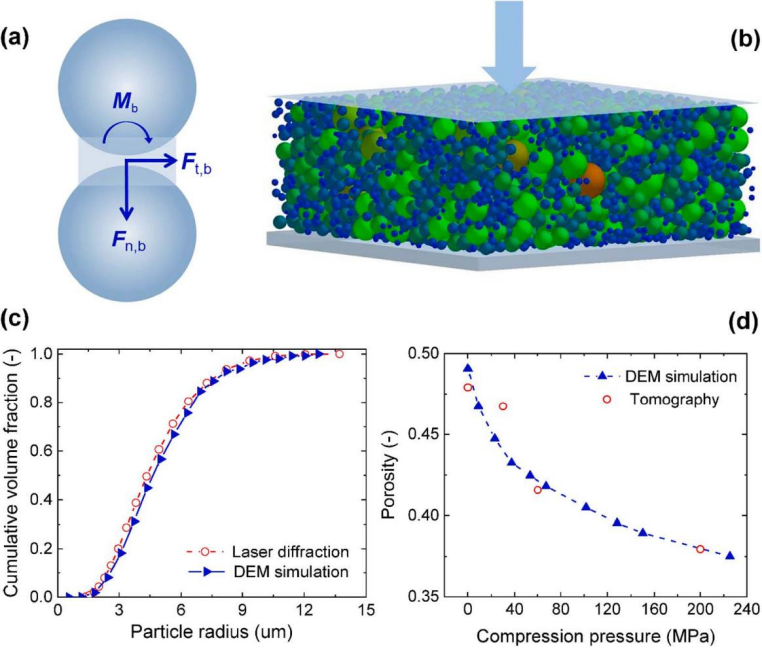
压延后概率分布向左移动,对应于平均球形度从 0.82 降低到 0.80。颗粒取向角定义为颗粒的最长轴与垂直于集电器的 z 轴间的角度,概率分布从 0° 到 45° 先逐渐增大,之后趋于平缓。对于压延结构,颗粒球形度的轻微下降和颗粒取向角的增加可归因于压延过程中颗粒变形和旋转的发生。
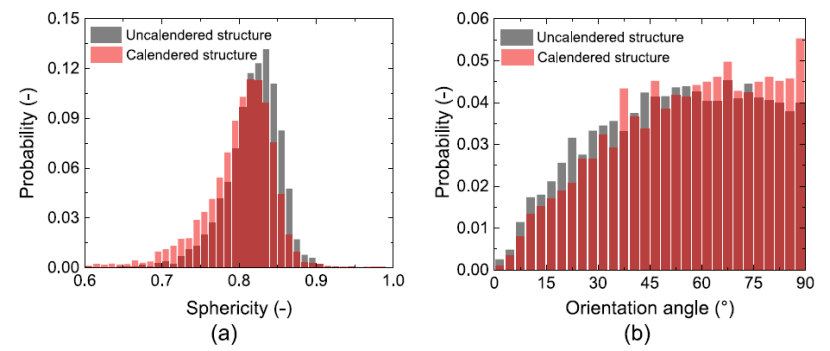
为解决颗粒形状和方向的影响,使用椭球近似描述断层扫描的颗粒堆积。采用连续孔径分布(c-PSD)和压汞孔径分布(MIP-PSD)两种计算方法,获得孔隙率分布曲线。显示出三个不同的阶段,高累积孔隙体积分数的初始阶段,随后迅速下降,最后以较低的斜率降至零。MIP 曲线比相应的 c-PSD 曲线下降得更剧烈。DEM 模拟和椭球结构与压延和断层扫描模型展现出相似的变化趋势。
对于50% 累积孔隙体积分数的孔隙半径值,断层扫描结构具有最小的孔径半径, DEM 模拟的球形颗粒孔径增加了不到 10%。结果表明,使用球形颗粒近似的 DEM 模拟可以捕捉压延过程中孔隙率分布的演变。
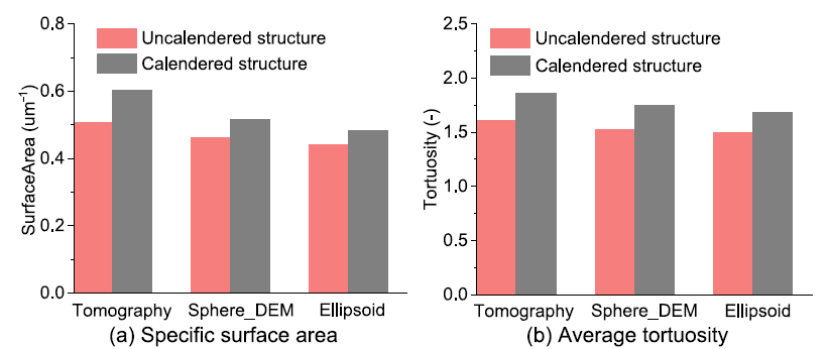
计算不同条件下未压延和压延结构的比表面积和迂曲度值,压延后表面积增加,DEM 模拟结构和椭球体结构的比表面积值略低于层析成像结构。对于平均曲折因子,断层扫描结构约为比 DEM 模拟结构和椭球结构高 5% 到 10%。压延后,电池电极的孔隙率降低,多孔相的“扭曲”增加。随颗粒堆积变得更密,比表面积增加,从而在界面处实现更好的 Li+ 离子扩散性能。
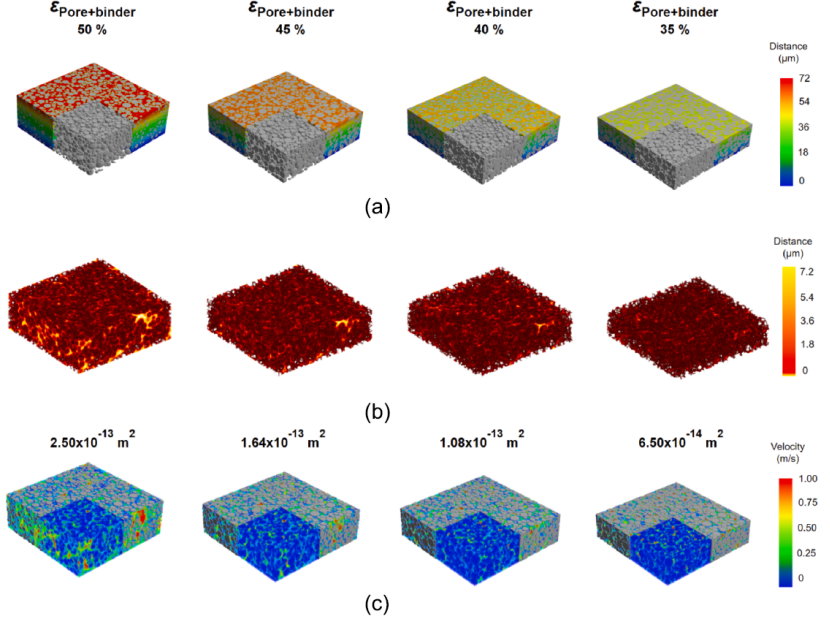
DEM 模拟过程中的微观结构演变如图所示,将 AM 颗粒结构转换为 3D 体素化图像,横截面尺寸为 200 × 200 μm。颗粒间隙随逐步压延减小,渗透率从 2.5×10-13 m2 降低到 6.5×10-14 m2 ,表明压延对孔隙相传输特性和微观结构非均质性的影响。
CBD 相生成和电化学分析
使用图像分割进一步分析断层扫描中的 CBD 相。压延和未压延结构的孔隙相体积分数分别为 11.52% 和 6.19%。应用粘合剂生成算法将等体积的 CBD 相添加到相应的 DEM 生成结构中

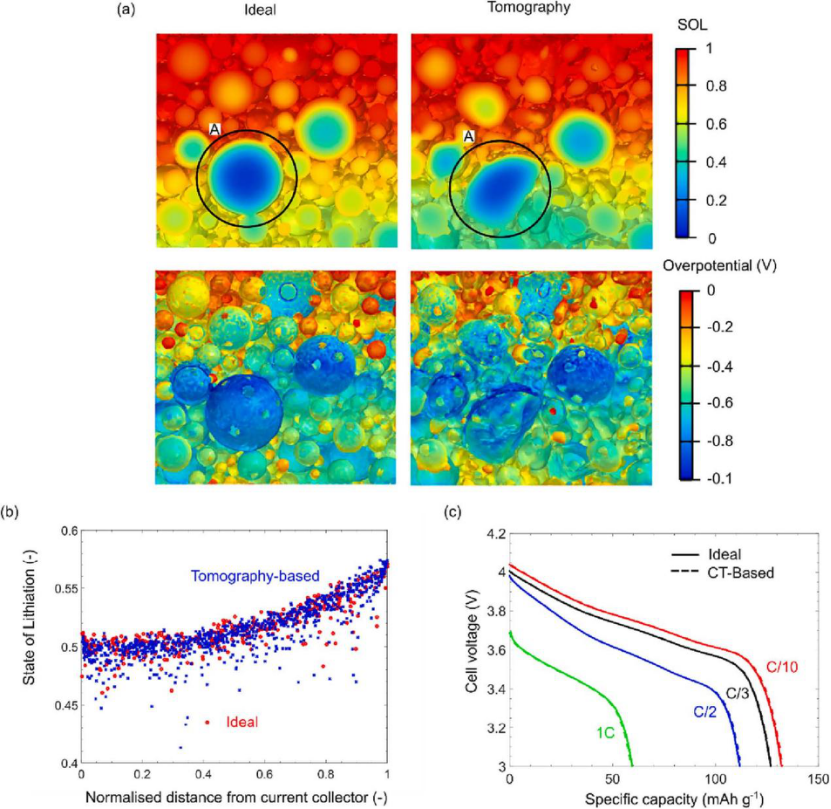
以定性的方式比较基于未压延断层扫描微观结构的锂化状态和过电势分布及其在 DEM 中使用的等效理想化情况。结果表明,理想化的几何形状足以捕捉电极微结构的整体性能。
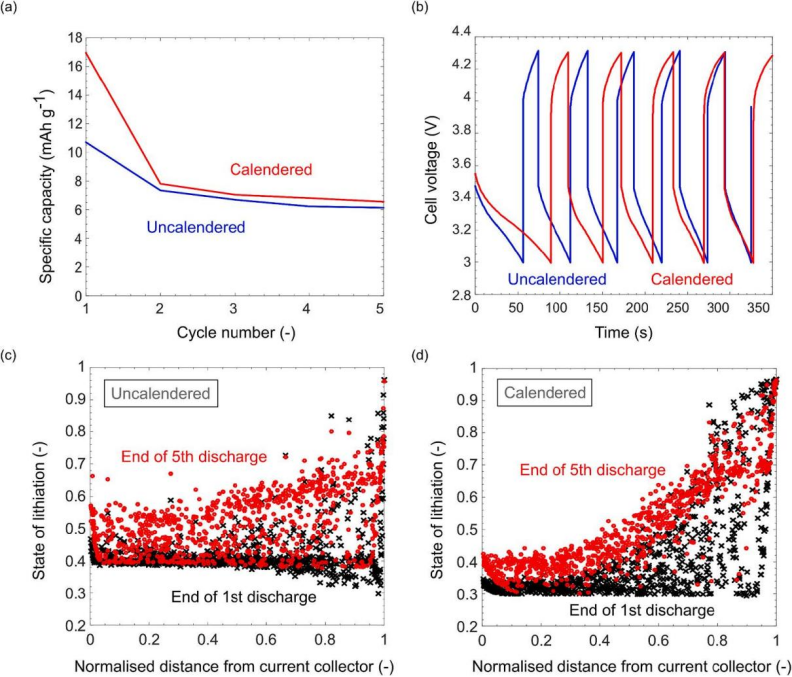
考虑对下一代电池电极开发至关重要的场景:循环条件下的高倍率充电和放电。以 5C 的速率进行 5 个循环,图a表明无论压延条件如何,放电容量在高倍率下都会显着下降,可归因于放电和充电结束时经历的大极化(图b)。明显地,压延电极的性能要好一些(图c-d),并且材料利用率有所提高。高倍率循环的研究强调了电极微观结构设计的必要性,特别是在涉及厚电极时。
研究表明锂离子电池的微观结构对其性能有显着影响。孔隙率为电解质渗透和锂离子传输提供曲折的路径,颗粒尺寸决定锂化发生的速率,碳粘合剂的水平影响电子传导。这三种机制以复杂的方式竞争并控制电极的性能,电极的倍率性能主要由这些机制决定,并且需要复杂的多参数优化。
结论
从数值建模的角度来看,压延过程非常复杂且计算要求较高。本研究提出一个包含 XCT 表征、DEM 和电化学测试的数字化工作流程,建立了压延工艺与微观结构性能之间的联系。高保真的 3D 物理模拟可以预测不同压延条件下的微观结构变化、颗粒尺度信息和相应的电池性能。未来的工作还将包括轧机速度、温度和压力等综合特征。
电极优化可以通过诸如对整个电极的孔隙分布进行分级等技术实现,XCT 和电化学模拟的结合可以更好地了解电极形态及其对电池性能的影响。本项目提出的方法将进一步用于研究考虑不同厚度和孔隙率的电极结构演变,为电极设计提供有价值的工具。
参考
- Ge R, Boyce A M, Zhang Y S, et al. Discrete element method and electrochemical modelling of lithium ion cathode structures characterised by X-ray computed tomography[J]. Chemical Engineering Journal, 2023, 465: 142749.