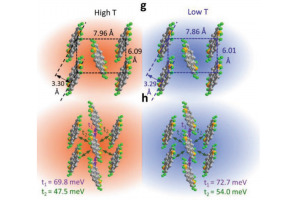
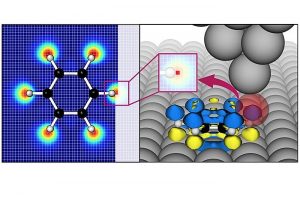
溶液处理的二维有机半导体在光电子、生物传感器方面的应用,是近年的研究热点。但是制备分子取向规则、缺陷密度低的二维有机晶体薄膜仍然面临挑战。香港大学冯宪平课题组与新墨西哥州立大学Paddy Kwok Leung Chan课题组使用超慢剪切法(Ultraslow Shearing Method)制备出高度结晶的C10-DNTT单层晶体。通过表面能计算,证实其动力学Wulff构造生长模式。 得到的无缝、高度结晶单层作为模板,在上面热沉积另一个超薄C10-DNTT结晶单层,这种方式制备的薄膜,其分子取向完全复制了模板的取向。C10-DNTT制备的有机场效应晶体管载流子迁移率可以达到14.7 cm2/V·s,而纯粹的热蒸发法只能达到7.3 cm2/V·s,溶液剪切法为2.8 cm2/V·s。这种简单有效的方法,可能用于大规模制备高性能、低成本电子产品。 载流子迁移率的计算,采用AMS软件中的ADF模块,通过转移积分的计算得到。 Zhiwen Zhou, Qisheng Wu, Sijia Wang, Yu-Ting Huang, Hua Guo, Shien-Ping Feng,* and Paddy Kwok Leung Chan*, Field-Effect Transistors Based on 2D Organic Semiconductors Developed by a Hybrid Deposition Method, Adv. Sci. 2019, 1900775