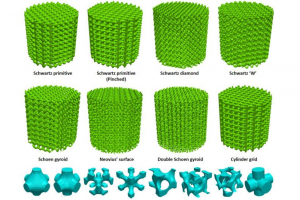
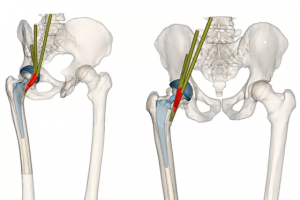
一、概述 抗生素耐药细菌种类的增加推动了对最小化感染风险医疗器械的需求。抗菌功能可以通过修改植入物的设计实现,即并入一个局部释放治疗剂的储存库,因此维持植入物的机械功能至关重要。本研究通过增材制造探索可能实现设计灵活性的机会,开发多种内部多孔晶格结构模型用于最大限度地提高装载药物体积,同时保持髋关节植入物的承载能力。 图1:髋关节植入物的 CAD设计(a)传统植入物(b)晶格设计提供装载治疗剂的孔隙空间 二、设计、模拟与制造 2.1 晶胞设计 目前二期髋关节翻修占位器的使用寿命有限且无法承受患者的全部负荷,本研究对其具有内部晶格的结构优化进行研究。晶格根据多孔金属压缩试验国际标准 ISO 13314 设计,选择压缩晶格直径 Do 和 高度 Ho 均为 15 mm的圆柱体试样,遵循 Ho = Do ~ 2 Do,试样直径与平均孔径 da 的关系为 Do ≥ 10 da。值得注意的是,M2 Cusing SLM 系统(德国 Concept Laser)的分辨率要求最小支柱直径约为 0.2 mm 才能达到可接受的零件质量。 使用 Simpleware 软件创建 8 种不同晶胞类型的压缩晶格结构。为能够加入最大体积的治疗剂,每种晶胞类型的目标体积分数(固体与孔隙的比率)以 10 为增量递减,直到在保持所有设计参数的情况下确定最低可能的体积分数。晶胞周期(沿轴填充图像域的晶胞数量)在 X、Y 和 Z 方向上相同,以 5 为步长变化。随着体积分数降低或晶胞周期增加,支柱直径会减小。因此,在确定可能的最低体积分数后,为每个压缩晶格设计最高的晶胞周期,这将为每种晶胞类型提供在 SLM 设备限制范围内的最小支柱直径。 2.2 有限元分析 在 Simpleware FE 中为每个晶格结构生成高质量的四面体网格模型,导入 Comsol Multiphysics 软件进行有限元分析。轴向施加 2300 N,底面施加所有方向的固定约束。设置 […]