粒子迁移率对于有机电子器件例如场致发射晶体管(OFET)、有机发光二极管、光伏电池非常关键。载流子从一个位置迁移到另一个位置,迁移率主要由转移积分决定。ADF可以直接计算转移积分。作为简化的例子,本例计算两个萘分子之间的电子迁移率。

粒子迁移率对于有机电子器件例如场致发射晶体管(OFET)、有机发光二极管、光伏电池非常关键。载流子从一个位置迁移到另一个位置,迁移率主要由转移积分决定。ADF可以直接计算转移积分。作为简化的例子,本例计算两个萘分子之间的电子迁移率。
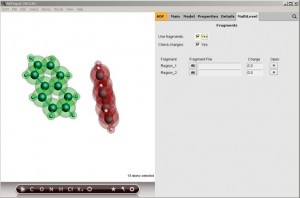
说明:氨基酸在水中通常会形成H键。因此不能使用通常的溶剂化模型如COSMO、SCRF等等来描述,而应该将溶剂分子直接地与溶质分子同等地考虑在内。但溶剂分子的热运动导致溶剂分子相对于溶质的位置比较难以确定。 精确的处理这类问题,需要进行第一性原理分子动力学或者第一性原理的蒙特卡洛模拟,得到溶剂分子相对于溶质分子的位置。这样工作量比较大。 一个比较节省的,可靠性也较高的方式,是将溶剂分子按可能形成氢键的位置预先摆放在溶质分子附近,通过几何结构优化,找到溶剂分子相对于溶质分子的位置。然后基于这样的几何结构计算原子的电荷分配。 我们以甘氨酸举例。
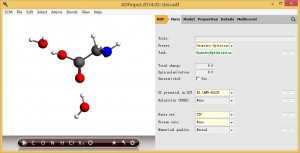
1,将优化好的结构导入ADFinput,并依次设置参数如下图所示:
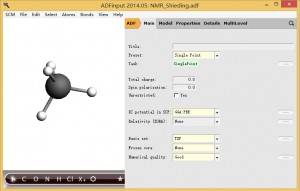
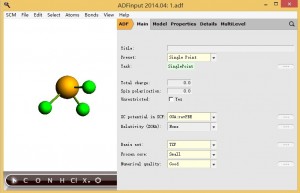
1,对分子进行几何结构优化 2,主面板参数设置如下(注意对于重元素应该另设置基组为TZ2P或QZ4P):
首先得到发生电荷转移的体系的总体结构,通常预先由用户给定,或通过几何结构优化得到。假设用户已经得到体系的结构,如下图所示:

1,使用MMC-SA优化ReaxFF参数 主讲人:Eldhose Iype 时 间:2014年12月2日 北京时间23:00 人 数:暂无限制
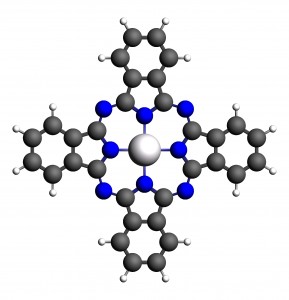
参考文献:Ground and Excited States of Zinc Phthalocyanine Studied by Density Functional Methods, G. Ricciardi and A. Rosa, E. J. Baerends, J. Phys. Chem. A 2001, 105, 5242-5254 锌酞菁本身具有荧光性,本文计算了锌酞菁的基态和激发态性质。基于前一篇文章的内容,我们此处主要介绍激发态计算、激发态结构优化、激发态频率计算、Franck-Condon因子计算等。 1, 采用优化好的锌酞菁分子结构进行计算: ADFinput支持比较灵活的分子拷贝、粘贴操作,例如: 将前一次计算的结构,从*.run文件
概述 Forcefield 模块包括超过种类丰富经验势,可以模拟的材料种类涵盖了金属、半导体合金、玻璃结构和有机材料等。此外,ForceField还支持机器学习力场 ML-MTPs,大大扩展了力场方法的应用范围。 更多功能介绍详见 QuantumATK功能列表。 力场(经验势)类型 ML-MTPs机器学习力场 超过 300 种键级势/力场 二体、三体势:多种版本的 Lennard-Jones、Coulomb、Stillinger-Weber、Tersoff、Brenner、Morse、Buckingham、Vessal、Tosi-Fumi,用户自定义 多体势:EAM、MEAM、Finnis-Sinclair、Sutton-Chen、charge-optmized many-body(COMB) 可极化势:Madden/Tangney-Scandolo,core-shell ReaxFF 和 ReaxFF+ (来自 AQcomputare) Valence force field(VFF)模型 BYOP(Bring Your Own Potential) Python 接口,用于添加以上任何支持类型的势(用户自己的参数或来自其他文献参数) 支持将几种力场结合 例如将 Stillinger-Weber 势与 Lennard-Jones 项结合来考虑范德华相互作用 包含了已见于文献的势:Pedone,Guillot-Sator,Marian-Gastreich,Feuston-Garofalini,Mastsui,Leinenweber,Madden 等 库仑作用解法 Ewald(平滑粒子网格),DSF,Debye,simple pairwise 并行化计算 与Forcefield配合,NanoLab提供的高级建模工具和性质分析工具为经验势分子动力学计算提供更多的便利。使用ATK-Forcefield进行分子动力学计算请参考:QuantumATK:更强大、更灵活的材料动力学模拟。 自定义势函数 ATK-Forcefield提供的图形界面工具BYOP(Bring Your Own Potential)支持自定义力场参数或力场组合。 支持几种势结合使用,例如将 Stillinger-Weber 势与 Lennard-Jones 势联合使用以考虑范德华相互作用 方便的支持文献中提到的势,例如 Pedone、Guillot-Sator、Marian-Gasteich、Feuston-Garofalini、Matsui、Leinenweber…… 各种势的细节参见手册 […]
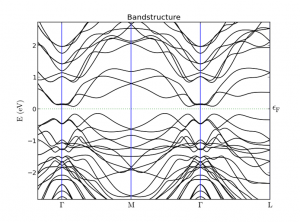
QuantumATK 中的 SemiEmpirical 通过对哈密顿量的参数化大大节省了计算量,可以用于更接近实验尺度的模拟。SemiEmpirical 中包含 DFTB 和 EHT 两种半经验哈密顿量计算方法。 更多功能介绍详见 QuantumATK功能列表。 DFTB 模型,QuantumATK 中提供30套参数,更多参数可以网站下载直接使用 内置 Slater-Koster 模型,内置第四族半导体和三五族化合物合金的参数模型 用户自定义 Slater-Koster 参数 扩展 Huckel 模型提供超过 300 种预定义的基组,用于周期表几乎全部元素 元素周期表中几乎所有元素的 Muller 和 Hoffmann 基组(用于分子计算) Cerda 参数,用于二元合金,半导体,金属,石墨(烯)等 带应变体系的紧束缚模型(T. B. Boykin et al., Phys. Rev. B 81, 125202 (2010)) 用内置的自旋分裂参数数据库增加自旋极化项 非共线自旋 自旋轨道耦合(参数化方法) 增加 Hartree 项,用来反映对静电场的自洽响应 所有模型都采用 DFTB 方法包含来自外部数据库的原子 Hartree 项自洽响应项,可以用来进行自洽计算 解析计算力和张力 DFTB/SK方法可以用于各种(分子、块体、器件)体系的多种计算: […]
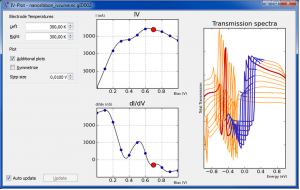
概述 DFT-LCAO实现了赝势和原子轨道线性组合(LCAO)相结合的第一性原理密度泛函理论电子结构计算方法,LCAO使用的数值原子轨道基组能够更方便地控制基组参数。ATK-DFT内置的模守恒赝势则涵盖了元素周期表中全部的元素。ATK-DFT实现了众多版本的局域密度近似(LDA)和广义梯度近似(GGA)的交换关联函数。meta-GGA、DFT-1/2则可以准确、快速地计算半导体、绝缘体材料的带隙。ATK-DFT支持Hubbard+U模型(LSDA+U, SGGA+U),可以更好地处理强关联体系。 更多功能介绍详见 QuantumATK功能列表。 基于密度泛函理论(DFT)和原子轨道线性组合(LCAO)基组的计算引擎数值轨道基组 对多数元素优化过的基组:https://molmod.ugent.be/deltacodesdft 模守恒 Troullier-Martins 赝势 FHI/SG15/PseudoDojo 势,用于周期表包括镧系在内的全部元素,很多元素支持半芯势PseudoDojo 和 SG15 势是全相对论的 超过 300 种 LDA/GGA/MGGA 交换相关泛函(libXC) LDA:HL,PW,PZ,RPA,WIGNER,XA 等GGA:BLYP,BP86,BPW91,PBE,PBES,PW91,RPBE,XLYP 等 杂化泛函支持HSE06、HSE06-DDH、B3LYP、B3LYP5、PBE3等与LCAO基组结合,计算高效、精确比平面波杂化泛函方法快上百倍可以计算超大体系(~2000原子)的性质用于半导体和绝缘体精确带隙计算的方法 MetaGGADFT+1/2经验的“赝势投影算符移动”(Pseudopotential Projector Shift,PPS)方法(内含 Si 和 Ge 的参数) 范德华力模型(DFT-D2 和 DFT-D3)非共线、限制性和非限制性的自旋极化计算自旋-轨道耦合Hubbard U 项( LDA 或 GGA),可以自旋区分 “Dual”,“on-site”,“shell-wise” 方法 Counterpoise 校正基组重叠误差(BSSE)Ghost 原子(真空基组),更精确的描述表面和空隙虚晶近似(Virtual Crystal Approximation,VCA)解析计算力和张力 DFT-LCAO的计算性能 计算精度 使用内嵌的最新赝势和泛函 DFT-LCAO,可以在能量与结构优化、电子态和半导体带隙等全方位的计算中获得最佳的计算精度。 固体结构SCAN泛函在计算能量和结构方面有巨大的优势,详见:SCAN metaGGA介绍;Delta测试结果表明,PseudoDojo赝势+LCAO基组对于全元素周期表的计算精度很好(~1 meV)。半导体带隙HSE06、MetaGGA、DFT+1/2、HSE、PPS等多种方法用于在计算半导体精确带隙。 计算速度 DFT-LCAO 对大体系的计算能力优势明显,可以高效完成平面波基组无法很难实现的数前原子体系的自洽计算,随着版本的更新,计算速度提升明显。DFT-LCAO […]