
概述
DFT-LCAO实现了赝势和原子轨道线性组合(LCAO)相结合的第一性原理密度泛函理论电子结构计算方法,LCAO使用的数值原子轨道基组能够更方便地控制基组参数。ATK-DFT内置的模守恒赝势则涵盖了元素周期表中全部的元素。ATK-DFT实现了众多版本的局域密度近似(LDA)和广义梯度近似(GGA)的交换关联函数。meta-GGA、DFT-1/2则可以准确、快速地计算半导体、绝缘体材料的带隙。ATK-DFT支持Hubbard+U模型(LSDA+U, SGGA+U),可以更好地处理强关联体系。
更多功能介绍详见 QuantumATK功能列表。
- 基于密度泛函理论(DFT)和原子轨道线性组合(LCAO)基组的计算引擎
- 数值轨道基组
- 对多数元素优化过的基组:https://molmod.ugent.be/deltacodesdft
- 模守恒 Troullier-Martins 赝势
- FHI/SG15/PseudoDojo 势,用于周期表包括镧系在内的全部元素,很多元素支持半芯势
- PseudoDojo 和 SG15 势是全相对论的
- 超过 300 种 LDA/GGA/MGGA 交换相关泛函(libXC)
- LDA:HL,PW,PZ,RPA,WIGNER,XA 等
- GGA:BLYP,BP86,BPW91,PBE,PBES,PW91,RPBE,XLYP 等
- 杂化泛函支持HSE06、HSE06-DDH、B3LYP、B3LYP5、PBE3等
- 与LCAO基组结合,计算高效、精确
- 比平面波杂化泛函方法快上百倍
- 可以计算超大体系(~2000原子)的性质
- 用于半导体和绝缘体精确带隙计算的方法
- MetaGGA
- DFT+1/2
- 经验的“赝势投影算符移动”(Pseudopotential Projector Shift,PPS)方法(内含 Si 和 Ge 的参数)
- 范德华力模型(DFT-D2 和 DFT-D3)
- 非共线、限制性和非限制性的自旋极化计算
- 自旋-轨道耦合
- Hubbard U 项( LDA 或 GGA),可以自旋区分
- “Dual”,“on-site”,“shell-wise” 方法
- Counterpoise 校正基组重叠误差(BSSE)
- Ghost 原子(真空基组),更精确的描述表面和空隙
- 虚晶近似(Virtual Crystal Approximation,VCA)
- 解析计算力和张力
DFT-LCAO的计算性能
计算精度
使用内嵌的最新赝势和泛函 DFT-LCAO,可以在能量与结构优化、电子态和半导体带隙等全方位的计算中获得最佳的计算精度。
- 固体结构
- SCAN泛函在计算能量和结构方面有巨大的优势,详见:SCAN metaGGA介绍;
- Delta测试结果表明,PseudoDojo赝势+LCAO基组对于全元素周期表的计算精度很好(~1 meV)。
- 半导体带隙
- HSE06、MetaGGA、DFT+1/2、HSE、PPS等多种方法用于在计算半导体精确带隙。
计算速度
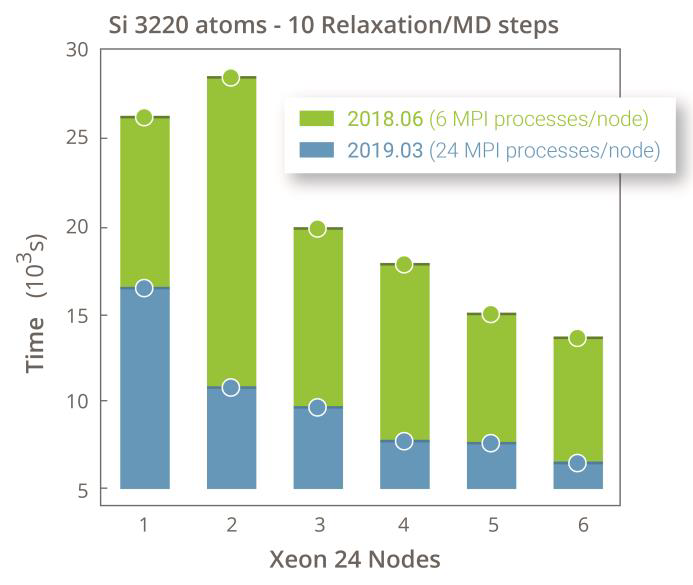
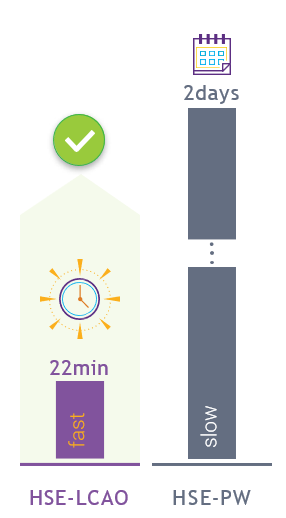
DFT-LCAO 对大体系的计算能力优势明显,可以高效完成平面波基组无法很难实现的数前原子体系的自洽计算,随着版本的更新,计算速度提升明显。DFT-LCAO 支持大规模的并行计算。

DFT-LCAO支持的计算类型
DFT-LCAO可以用于以下模型的计算:
- 分子(Molecule)
- 块体材料(Bulk)
- 器件或界面体系(Device)
- 表面体系(Suface)
DFT-LCAO可以进行各种性质的计算:
- 几何结构优化
- 分子动力学计算
- 结构、电子、光学等性质计算
- NEB过渡态与反应路径搜索
- 声子与热性质
- 电声耦合