
概述
Forcefield 模块包括超过种类丰富经验势,可以模拟的材料种类涵盖了金属、半导体合金、玻璃结构和有机材料等。此外,ForceField还支持机器学习力场 ML-MTPs,大大扩展了力场方法的应用范围。
更多功能介绍详见 QuantumATK功能列表。
力场(经验势)类型
- ML-MTPs机器学习力场
- 超过 300 种键级势/力场
- 二体、三体势:多种版本的 Lennard-Jones、Coulomb、Stillinger-Weber、Tersoff、Brenner、Morse、Buckingham、Vessal、Tosi-Fumi,用户自定义
- 多体势:EAM、MEAM、Finnis-Sinclair、Sutton-Chen、charge-optmized many-body(COMB)
- 可极化势:Madden/Tangney-Scandolo,core-shell
- ReaxFF 和 ReaxFF+ (来自 AQcomputare)
- Valence force field(VFF)模型
- BYOP(Bring Your Own Potential)
- Python 接口,用于添加以上任何支持类型的势(用户自己的参数或来自其他文献参数)
- 支持将几种力场结合
- 例如将 Stillinger-Weber 势与 Lennard-Jones 项结合来考虑范德华相互作用
- 包含了已见于文献的势:Pedone,Guillot-Sator,Marian-Gastreich,Feuston-Garofalini,Mastsui,Leinenweber,Madden 等
- 库仑作用解法
- Ewald(平滑粒子网格),DSF,Debye,simple pairwise
- 并行化计算


与Forcefield配合,NanoLab提供的高级建模工具和性质分析工具为经验势分子动力学计算提供更多的便利。使用ATK-Forcefield进行分子动力学计算请参考:QuantumATK:更强大、更灵活的材料动力学模拟。
自定义势函数
ATK-Forcefield提供的图形界面工具BYOP(Bring Your Own Potential)支持自定义力场参数或力场组合。
- 支持几种势结合使用,例如将 Stillinger-Weber 势与 Lennard-Jones 势联合使用以考虑范德华相互作用
- 方便的支持文献中提到的势,例如 Pedone、Guillot-Sator、Marian-Gasteich、Feuston-Garofalini、Matsui、Leinenweber……
- 各种势的细节参见手册
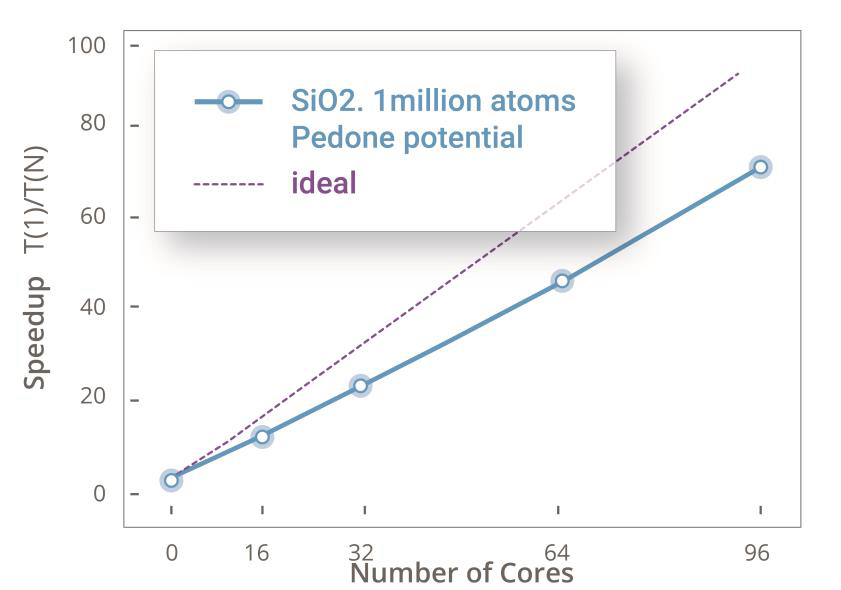
计算速度与并行化
当前 Forcefield 动力学可以使用MPI/OpenMP或混合并行,大规模并行提速非常明显。
Forcefield的优势
- 比传统 MD 代码(LAMMPS 等)更灵活
- 方便定义自己的势和混合势
- 可以进行 NEGF 热输运计算
- 图形用户界面可以用于建模、计算设置和结果分析
立即试用 QuantumATK!
附:Forcefield中包含的经验势(力场)参数列表
更多信息参见:ForceField中包含的力场详细说明(英文)
TremoloX potential parameter sets
- Anwar_NaCl_2003 (Na, Cl)
- Jamshed Anwar and Daan Frenkel and Massimo G. Noro, Calculation of the melting point of NaCl by molecular simulation, The Journal of Chemical Physics, 118, pp. 728-735, 2003 link
- Billeter_HNOSi_2006 (H, Si, O, N)
- Billeter, Salomon R. and Curioni, Alessandro and Fischer, Dominik and Andreoni, Wanda, Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon, Phys. Rev. B, 73, p. 155329, 2006 link
- Broglia_HfOSi_2014 (Si, Hf, O)
- G Broglia and G Ori and L Larcher and M Montorsi, Molecular dynamics simulation of amorphous HfO2 for resistive RAM applications, Modelling and Simulation in Materials Science and Engineering, 22, p. 065006, 2014 link
- COMB_NTi_2014 (N, Ti)
- Y-T Cheng and T Liang and J A Martinez and S R Phillpot and S B Sinnott, A charge optimized many-body potential for titanium nitride (TiN), Journal of Physics: Condensed Matter, 26, p. 265004, 2014 link
- COMB_OSi_2007 (Si, O)
- Yu, Jianguo and Sinnott, Susan B. and Phillpot, Simon R., Charge optimized many-body potential for the Si/SiO2 system, Phys. Rev. B, 75, p. 085311, 2007 link
- COMB_OSi_2010 (Si, O)
- Shan, Tzu-Ray and Devine, Bryce D. and Hawkins, Jeffery M. and Asthagiri, Aravind and Phillpot, Simon R. and Sinnott, Susan B., Second-generation charge-optimized many-body potential for Si/SiO2 and amorphous silica, Phys. Rev. B, 82, p. 235302, 2010 link
- EAMFS_Ag_1987 (Ag)
- Ackland, GJ and Tichy, G and Vitek, V and Finnis, MW, Simple N-body potentials for the noble metals and nickel, Philosophical Magazine A, 56, pp. 735-756, 1987
- EAMFS_AlFe_2005 (Al, Fe)
- Mendelev and D. J. Srolovitz and G. J. Ackland and S. Han, Effect of Fe segregation on the migration of a non-symmetric Sigma 5 tilt grain boundary in Al, J. Mater. Res., 20, pp. 208-218, 2005
- EAMFS_AlMg_2009 (Mg, Al)
- Mendelev, MI and Asta, M and Rahman, MJ and Hoyt, JJ, Development of interatomic potentials appropriate for simulation of solid–liquid interface properties in Al–Mg alloys, Philosophical Magazine, 89, pp. 3269-3285, 2009
- EAMFS_AlSm_2015 (Al, Sm)
- Mendelev, MI and Zhang, F and Ye, Z and Sun, Y and Nguyen, MC and Wilson, SR and Wang, CZ and Ho, KM, Development of interatomic potentials appropriate for simulation of devitrification of Al90Sm10 alloy, Modelling and Simulation in Materials Science and Engineering, 23, p. 045013, 2015
- EAMFS_Al_2000 (Al)
- Sturgeon, Jess B and Laird, Brian B, Adjusting the melting point of a model system via Gibbs-Duhem integration: Application to a model of aluminum, Physical Review B, 62, p. 14720, 2000
- EAMFS_Al_2008 (Al)
- Mendelev, MI and Kramer, MJ and Becker, CA and Asta, M, Analysis of semi-empirical interatomic potentials appropriate for simulation of crystalline and liquid Al and Cu, Philosophical Magazine, 88, pp. 1723-1750, 2008
- EAMFS_Au_1987 (Au)
- Ackland, GJ and Tichy, G and Vitek, V and Finnis, MW, Simple N-body potentials for the noble metals and nickel, Philosophical Magazine A, 56, pp. 735-756, 1987
- EAMFS_CFe_2008 (C, Fe)
- Hepburn, Derek J and Ackland, Graeme J, Metallic-covalent interatomic potential for carbon in iron, Physical Review B, 78, p. 165115, 2008
- EAMFS_CuZr_2007 (Cu, Zr)
- Mendelev, MI and Sordelet, DJ and Kramer, MJ, Using atomistic computer simulations to analyze x-ray diffraction data from metallic glasses, Journal of Applied Physics, 102, pp. 043501-043501, 2007
- EAMFS_CuZr_2009 (Zr, Cu)
- Mendelev, MI and Kramer, MJ and Ott, RT and Sordelet, DJ and Yagodin, D and Popel, P, Development of suitable interatomic potentials for simulation of liquid and amorphous Cu–Zr alloys, Philosophical Magazine, 89, pp. 967-987, 2009
- EAMFS_Cu_1987 (Cu)
- Ackland, GJ and Tichy, G and Vitek, V and Finnis, MW, Simple N-body potentials for the noble metals and nickel, Philosophical Magazine A, 56, pp. 735-756, 1987
- EAMFS_Cu_2008 (Cu)
- Mendelev, MI and Kramer, MJ and Becker, CA and Asta, M, Analysis of semi-empirical interatomic potentials appropriate for simulation of crystalline and liquid Al and Cu, Philosophical Magazine, 88, pp. 1723-1750, 2008
- EAMFS_FeP_2004 (P, Fe)
- Ackland, GJ and Mendelev, MI and Srolovitz, DJ and Han, S and Barashev, AV, Development of an interatomic potential for phosphorus impurities in alpha-iron, Journal of Physics: Condensed Matter, 16, p. S2629, 2004
- EAMFS_FeV_2007 (Fe, V)
- Mendelev, Mikhail I and Han, Seungwu and Son, Won-joon and Ackland, Graeme J and Srolovitz, David J, Simulation of the interaction between Fe impurities and point defects in V, Physical Review B, 76, p. 214105, 2007
- EAMFS_Fe_1997 (Fe)
- Ackland, GJ and Bacon, DJ and Calder, AF and Harry, T, Computer simulation of point defect properties in dilute Fe—Cu alloy using a many-body interatomic potential, Philosophical Magazine A, 75, pp. 713-732, 1997
- EAMFS_Fe_2003 (Fe)
- Mendelev, MI and Han, S and Srolovitz, DJ and Ackland, GJ and Sun, DY and Asta, M, Development of new interatomic potentials appropriate for crystalline and liquid iron, Philosophical magazine, 83, pp. 3977-3994, 2003
- EAMFS_Fe_2003b (Fe)
- Mendelev, MI and Han, S and Srolovitz, DJ and Ackland, GJ and Sun, DY and Asta, M, Development of new interatomic potentials appropriate for crystalline and liquid iron, Philosophical magazine, 83, pp. 3977-3994, 2003
- EAMFS_Mg_2006 (Mg)
- Sun, DY and Mendelev, MI and Becker, CA and Kudin, K and Haxhimali, Tomorr and Asta, M and Hoyt, JJ and Karma, A and Srolovitz, DJ, Crystal-melt interfacial free energies in hcp metals: A molecular dynamics study of Mg, Physical Review B, 73, p. 024116, 2006
- EAMFS_Na_2015 (Na)
- Wilson, SR and Gunawardana, KGSH and Mendelev, MI, Solid-liquid interface free energies of pure bcc metals and B2 phases, The Journal of chemical physics, 142, p. 134705, 2015
- EAMFS_NiZr_2012 (Ni, Zr)
- Mendelev, MI and Kramer, MJ and Hao, SG and Ho, KM and Wang, CZ, Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy, Philosophical Magazine, 92, pp. 4454-4469, 2012
- EAMFS_NiZr_2015 (Ni, Zr)
- Wilson, SR and Mendelev, MI, Anisotropy of the solid–liquid interface properties of the Ni–Zr B33 phase from molecular dynamics simulation, Philosophical Magazine, 95, pp. 224-241, 2015
- EAMFS_Ni_1987 (Ni)
- Ackland, GJ and Tichy, G and Vitek, V and Finnis, MW, Simple N-body potentials for the noble metals and nickel, Philosophical Magazine A, 56, pp. 735-756, 1987
- EAMFS_Ni_2012 (Ni)
- Mendelev, MI and Kramer, MJ and Hao, SG and Ho, KM and Wang, CZ, Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy, Philosophical Magazine, 92, pp. 4454-4469, 2012
- EAMFS_Ru_2008 (Ru)
- Fortini, Andrea and Mendelev, Mikhail I and Buldyrev, Sergey and Srolovitz, David, Asperity contacts at the nanoscale: Comparison of Ru and Au, Journal of Applied Physics, 104, pp. 074320-074320, 2008
- EAMFS_Ti_1992 (Ti)
- Ackland, Graeme J, Theoretical study of titanium surfaces and defects with a new many-body potential, Philosophical Magazine A, 66, pp. 917-932, 1992
- EAM_AgCu_2006 (Ag, Cu)
- Williams, PL and Mishin, Y and Hamilton, JC, An embedded-atom potential for the Cu–Ag system, Modelling and Simulation in Materials Science and Engineering, 14, p. 817, 2006
- EAM_AgCu_2009 (Ag, Cu)
- Wu, Henry H and Trinkle, Dallas R, Cu/Ag EAM potential optimized for heteroepitaxial diffusion from ab initio data, Computational Materials Science, 47, pp. 577-583, 2009
- EAM_AgHPd_Hybrid_2013 (H, Pd, Ag)
- Hale, Lucas Michael and Wong, Bryan Matthew and Zimmerman, Jonathan A and Zhou, XW, Atomistic potentials for palladium–silver hydrides, Modelling and Simulation in Materials Science and Engineering, 21, p. 045005, 2013
- EAM_AgHPd_Morse_2013 (H, Pd, Ag)
- Hale, Lucas Michael and Wong, Bryan Matthew and Zimmerman, Jonathan A and Zhou, XW, Atomistic potentials for palladium–silver hydrides, Modelling and Simulation in Materials Science and Engineering, 21, p. 045005, 2013
- EAM_Ag_2004 (Ag)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Ag_2006 (Ag)
- Williams, PL and Mishin, Y and Hamilton, JC, An embedded-atom potential for the Cu–Ag system, Modelling and Simulation in Materials Science and Engineering, 14, p. 817, 2006
- EAM_Ag_Sheng_2011 (Ag)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_AlAg_Sheng_2011 (Al, Ag)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_AlCu_Sheng_2011 (Al, Cu)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_AlMg_1997 (Mg, Al)
- Liu, Xiang-Yang and Ohotnicky, PP and Adams, JB and Rohrer, C Lane and Hyland Jr, RW, Anisotropic surface segregation in Al Mg alloys, Surface science, 373, pp. 357-370, 1997
- EAM_AlMnPd_2012 (Mn, Al, Pd)
- Schopf, Daniel and Brommer, Peter and Frigan, Benjamin and Trebin, Hans-Rainer, Embedded atom method potentials for Al-Pd-Mn phases, Physical Review B, 85, p. 054201, 2012
- EAM_AlNbTi_1996 (Nb, Al, Ti)
- Farkas, Diana and Jones, Chris, Interatomic potentials for ternary Nb-Ti-Al alloys, Modelling and Simulation in Materials Science and Engineering, 4, p. 23, 1996
- EAM_AlNiH_1997 (H, Al, Ni)
- Baskes, MI and Sha, Xianwei and Angelo, JE and Moody, NR, Trapping of hydrogen to lattice defects in nickel, Modelling and Simulation in Materials Science and Engineering, 5, p. 651, 1997Angelo, James E and Moody, Neville R and Baskes, Michael I, Trapping of hydrogen to lattice defects in nickel, Modelling and Simulation in Materials Science and Engineering, 3, p. 289, 1995
- EAM_AlNi_2002 (Ni, Al)
- Mishin, Y and Mehl, MJ and Papaconstantopoulos, DA, Embedded-atom potential for B2-NiAl, Physical Review B, 65, p. 224114, 2002
- EAM_AlNi_2004 (Ni, Al)
- Mishin, Yuri, Atomistic modeling of the gamma and gamma’-phases of the Ni–Al system, Acta materialia, 52, pp. 1451-1467, 2004
- EAM_AlNi_2009 (Ni, Al)
- Purja Pun, GP and Mishin, Y, Development of an interatomic potential for the Ni-Al system, Philosophical Magazine, 89, pp. 3245-3267, 2009
- EAM_AlPb_2000 (Pb, Al)
- Landa, A and Wynblatt, P and Siegel, DJ and Adams, JB and Mryasov, ON and Liu, X-Y, Development of glue-type potentials for the Al–Pb system: phase diagram calculation, Acta materialia, 48, pp. 1753-1761, 2000
- EAM_AlTi_2003 (Al, Ti)
- Zope, Rajendra R and Mishin, Yu, Interatomic potentials for atomistic simulations of the Ti-Al system, Physical Review B, 68, p. 024102, 2003
- EAM_AlZr_Sheng_2011 (Al, Zr)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Al_1999 (Al)
- Mishin, Y and Farkas, D and Mehl, MJ and Papaconstantopoulos, DA, Interatomic potentials for monoatomic metals from experimental data and ab initio calculations, Physical Review B, 59, p. 3393, 1999
- EAM_Al_2003 (Al)
- Zope, Rajendra R and Mishin, Yu, Interatomic potentials for atomistic simulations of the Ti-Al system, Physical Review B, 68, p. 024102, 2003
- EAM_Al_2009 (Al)
- Winey, JM and Kubota, Alison and Gupta, YM, A thermodynamic approach to determine accurate potentials for molecular dynamics simulations: thermoelastic response of aluminum, Modelling and Simulation in Materials Science and Engineering, 17, p. 055004, 2009
- EAM_Al_Lui_2004 (Al)
- Liu, Xiang-Yang and Ercolessi, Furio and Adams, James B, Aluminium interatomic potential from density functional theory calculations with improved stacking fault energy, Modelling and Simulation in Materials Science and Engineering, 12, p. 665, 2004
- EAM_Al_Sheng_2011 (Al)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Al_Zhou_2004 (Al)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Au_2004 (Au)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Au_2005 (Au)
- Grochola, Gregory and Russo, Salvy P and Snook, Ian K, On fitting a gold embedded atom method potential using the force matching method, The Journal of chemical physics, 123, p. 204719, 2005
- EAM_Au_Sheng_2011 (Au)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Ca_Sheng_2011 (Ca)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Ce_Sheng_2011 (Ce)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Co_2004 (Co)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Co_2012 (Co)
- Pun, GP Purja and Mishin, Y, Embedded-atom potential for hcp and fcc cobalt, Physical Review B, 86, p. 134116, 2012
- EAM_CrFeNi_2013 (Cr, Fe, Ni)
- Bonny, G and Castin, N and Terentyev, D, Interatomic potential for studying ageing under irradiation in stainless steels: the FeNiCr model alloy, Modelling and Simulation in Materials Science and Engineering, 21, p. 085004, 2013
- EAM_CuFeNi_2009 (Ni, Fe, Cu)
- Bonny, Giovanni and Pasianot, Roberto C and Castin, Nicolas and Malerba, Lorenzo, Ternary Fe–Cu–Ni many-body potential to model reactor pressure vessel steels: First validation by simulated thermal annealing, Philosophical Magazine, 89, pp. 3531-3546, 2009
- EAM_CuZr_Sheng_2011 (Zr, Cu)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Cu_2001b (Cu)
- Mishin, Yu and Mehl, MJ and Papaconstantopoulos, DA and Voter, AF and Kress, JD, Structural stability and lattice defects in copper: Ab initio, tight-binding, and embedded-atom calculations, Physical Review B, 63, p. 224106, 2001
- EAM_Cu_2004 (Cu)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Cu_Sheng_2011 (Cu)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_FeNi_2009 (Ni, Fe)
- Bonny, Giovanni and Pasianot, RC and Malerba, Lorenzo, Fe–Ni many-body potential for metallurgical applications, Modelling and Simulation in Materials Science and Engineering, 17, p. 025010, 2009
- EAM_Fe_2004 (Fe)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_HPd_2008 (H, Pd)
- Zhou, XW and Zimmerman, JA and Wong, BM and Hoyt, JJ, An embedded-atom method interatomic potential for Pd–H alloys, Journal of Materials Research, 23, pp. 704-718, 2008
- EAM_Ir_Sheng_2011 (Ir)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_MgCu_Sheng_2011 (Mg, Cu)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_MgTi_Sheng_2011 (Mg, Ti)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_MgY_Sheng_2011 (Y, Mg)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Mg_2004 (Mg)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_MoUXe_2013 (Mo, U, Xe)
- Smirnova, DE and Kuksin, A Yu and Starikov, SV and Stegailov, VV and Insepov, Z and Rest, J and Yacout, AM, A ternary EAM interatomic potential for U–Mo alloys with xenon, Modelling and Simulation in Materials Science and Engineering, 21, pp. 35011-35034, 2013
- EAM_Mo_2004 (Mo)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Nb_2010 (Nb)
- Fellinger, Michael R and Park, Hyoungki and Wilkins, John W, Force-matched embedded-atom method potential for niobium, Physical Review B, 81, p. 144119, 2010
- EAM_NiP_Sheng_2011 (Ni, P)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_NiZr_Sheng_2011 (Ni, Zr)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Ni_1999 (Ni)
- Mishin, Y and Farkas, D and Mehl, MJ and Papaconstantopoulos, DA, Interatomic potentials for monoatomic metals from experimental data and ab initio calculations, Physical Review B, 59, p. 3393, 1999
- EAM_Ni_2004 (Ni)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Ni_Sheng_2011 (Ni)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Pb_2004 (Pb)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Pb_Sheng_2011 (Pb)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_PdSi_Sheng_2011 (Si, Pd)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Pd_Sheng_2011 (Pd)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Pd_Zhou_2004 (Pd)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Pt_2004 (Pt)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Pt_Sheng_2011 (Pt)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Rh_Sheng_2011 (Rh)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Sr_Sheng_2011 (Sr)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Ta1_2013 (Ta)
- Ravelo, R and Germann, TC and Guerrero, O and An, Q and Holian, BL, Shock-induced plasticity in tantalum single crystals: Interatomic potentials and large-scale molecular-dynamics simulations, Physical Review B, 88, p. 134101, 2013
- EAM_Ta2_2013 (Ta)
- Ravelo, R and Germann, TC and Guerrero, O and An, Q and Holian, BL, Shock-induced plasticity in tantalum single crystals: Interatomic potentials and large-scale molecular-dynamics simulations, Physical Review B, 88, p. 134101, 2013
- EAM_Ta_2003 (Ta)
- Li, Youhong and Siegel, Donald J and Adams, James B and Liu, Xiang-Yang, Embedded-atom-method tantalum potential developed by the force-matching method, Physical Review B, 67, p. 125101, 2003
- EAM_Ta_2004 (Ta)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Ta_Sheng_2011 (Ta)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Ti_2004 (Ti)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_U_2013 (U)
- Smirnova, DE and Starikov, SV and Stegailov, VV, Interatomic potential for uranium in a wide range of pressures and temperatures, Journal of Physics: Condensed Matter, 24, p. 015702, 2012
- EAM_WHHe_Bonny_1_2014 (H, W, He)
- Bonny, Giovanni and Grigorev, Petr and Terentyev, Dmitry, On the binding of nanometric hydrogen–helium clusters in tungsten, Journal of Physics: Condensed Matter, 26, p. 485001, 2014
- EAM_WHHe_Bonny_2_2014 (H, W, He)
- Bonny, Giovanni and Grigorev, Petr and Terentyev, Dmitry, On the binding of nanometric hydrogen–helium clusters in tungsten, Journal of Physics: Condensed Matter, 26, p. 485001, 2014
- EAM_W_2004 (W)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Zhou_2004 (Ni, Mg, Co, Ag, Pt, W, Mo, Al, Pb, Zr, Au, Fe, Pd, Ti, Cu, Ta)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_ZrCuAg_Sheng_2011 (Ag, Cu, Zr)
- Fujita, T and Guan, PF and Sheng, HW and Inoue, A and Sakurai, T and Chen, MW, Coupling between chemical and dynamic heterogeneities in a multicomponent bulk metallic glass, Physical Review B, 81, p. 140204, 2010Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_ZrCuAl_Sheng_2011 (Cu, Zr, Al)
- Cheng, YQ and Ma, E and Sheng, HW, Atomic level structure in multicomponent bulk metallic glass, Physical review letters, 102, p. 245501, 2009Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_ZrPt_Sheng_2011 (Pt, Zr)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- EAM_Zr_2004 (Zr)
- Zhou, XW and Johnson, RA and Wadley, HNG, Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers, Physical Review B, 69, p. 144113, 2004
- EAM_Zr_Sheng_2011 (Zr)
- Sheng, HW and Kramer, MJ and Cadien, A and Fujita, T and Chen, MW, Highly optimized embedded-atom-method potentials for fourteen fcc metals, Physical Review B, 83, p. 134118, 2011
- FeustonGarofalini_CaHOSi_2004 (H, Ca, Al, O, Si, Na)
- Dolado, Jorge S and Griebel, Michael and Hamaekers, Jan, A molecular dynamic study of cementitious calcium silicate hydrate (C–S–H) gels, Journal of the American Ceramic Society, 90, pp. 3938-3942, 2007Litton, David A and Garofalini, Stephen H, Modeling of hydrophilic wafer bonding by molecular dynamics simulations, Journal of Applied Physics, 89, pp. 6013-6023, 2001Feuston, BP and Garofalini, SH, Onset of polymerization in silica sols, Chemical physics letters, 170, pp. 264-270, 1990Su, Xiaotao and Garofalini, Stephen H, Role of nitrogen on the atomistic structure of the intergranular film in silicon nitride: A molecular dynamics study, Journal of materials research, 19, pp. 3679-3687, 2004
- GuillotSator_OSiTiAlFe2MgCaNaK_2006 (Mg, Na, Ca, Al, O, Si, Fe, Ti, K)
- Bertrand Guillot and Nicolas Sator, A computer simulation study of natural silicate melts. Part I: Low pressure properties, Geochimica et Cosmochimica Acta, 71, pp. 1249 – 1265, 2007 link
- GuillotSator_OSiTiAlFe3MgCaNaK_2006 (Mg, Na, Ca, Al, O, Si, Fe, Ti, K)
- Bertrand Guillot and Nicolas Sator, A computer simulation study of natural silicate melts. Part I: Low pressure properties, Geochimica et Cosmochimica Acta, 71, pp. 1249 – 1265, 2007 link
- Guillot_KNCMFAT_2007 (Mg, Na, Ca, Al, O, Si, Fe, Ti, K)
- Bertrand Guillot and Nicolas Sator, A computer simulation study of natural silicate melts. Part I: Low pressure properties, Geochimica et Cosmochimica Acta, 71, pp. 1249 – 1265, 2007 link
- Iwasaki_AlCuNOSiTiW_2001 (Al, O, N, Si, W, Ti, Cu)
- Iwasaki,T. and Miura,H., Molecular dynamics analysis of adhesion strength of interfaces between thin films, Journal of Materials Research, 16, pp. 1789-1794, 2001
- JacksonCatlow_AlOSi_1988 (Si, Al, O, O_shell)
- Jackson, R. A. and Catlow, C. R. A., Computer Simulation Studies of Zeolite Structure, Molecular Simulation,, 1, pp. 207-224, 1988
- Jackson_HfO_2015 (Hf, O, O_shell)
- Araujo, R.M. and Valerio, M.E.G. and Jackson, R.A., Computer modelling of hafnium doping in lithium niobate, ArXiv e-prints, http://adsabs.harvard.edu/abs/2015arXiv150501661A
- Jackson_LiNbO_2015 (Nb, O_shell, O, Li)
- Araujo, R.M. and Valerio, M.E.G. and Jackson, R.A., Computer modelling of hafnium doping in lithium niobate, ArXiv e-prints, http://adsabs.harvard.edu/abs/2015arXiv150501661A
- Jahn_AlCaMgOSi_2007 (Ca, Si, Mg, O, Al)
- Sandro Jahn and Paul A. Madden, Modeling Earth materials from crustal to lower mantle conditions: A transferable set of interaction potentials for the CMAS system, Physics of the Earth and Planetary Interiors, 162, pp. 129 – 139, 2007 link
- Kerisit_LiOTi3_2010 (O_shell, Li, O, Ti)
- Kerisit, Sebastien and Rosso, Kevin M. and Yang, Zhenguo and Liu, Jun, Computer Simulation of the Phase Stabilities of Lithiated TiO2 Polymorphs, The Journal of Physical Chemistry C, 114, pp. 19096-19107, 2010
- Kerisit_LiOTi4_2010 (O_shell, Li, O, Ti)
- Kerisit, Sebastien and Rosso, Kevin M. and Yang, Zhenguo and Liu, Jun, Computer Simulation of the Phase Stabilities of Lithiated TiO2 Polymorphs, The Journal of Physical Chemistry C, 114, pp. 19096-19107, 2010
- Leinenweber_MgSiO_1988 (Mg, Si, O)
- Leinenweber, Kurt and Navrotsky, Alexandra, A transferable interatomic potential for crystalline phases in the system MgO—SiO2, Physics and Chemistry of Minerals, 15, pp. 588-596, 1988 link
- Lusvardi_SiPNaCaOF_2008 (F, Na, Ca, O, P, Si)
- Lusvardi, G and Malavasi, G and Cortada, M and Menabue, L and Menziani, M C and Pedone, A and Segre, U, Elucidation of the structural role of fluorine in potentially bioactive glasses by experimental and computational investigation., J Phys Chem B, 112, pp. 12730-9, 2008 link
- MEAM_SiOAu_2005 (Si, Au, O)
- Kuo, Chin-Lung and Clancy, Paulette, Development of atomistic MEAM potentials for the silicon–oxygen–gold ternary system, Modelling and Simulation in Materials Science and Engineering, 13, p. 1309, 2005
- MarianGastreich_SiBNH_2000 (H, Si, B, N)
- Marian, Christel M and Gastreich, Marcus, A systematic theoretical study of molecular Si/N, B/N, and Si/B/N (H) compounds and parameterisation of a force-field for molecules and solids, Journal of Molecular Structure: THEOCHEM, 506, pp. 107-129, 2000
- MarianGastreich_SiBN_2003 (Si, B, N)
- Gastreich, Marcus and Gale, Julian D and Marian, Christel M, Charged-particle potential for boron nitrides, silicon nitrides, and borosilazane ceramics: Derivation of parameters and probing of capabilities, Physical Review B, 68, p. 094110, 2003
- Marrocchelli_GeO_2010 (Ge, O)
- Marrocchelli, D. and Salanne, M. and Madden, P.A. and Simon, C. and Turq, P., The construction of a reliable potential for GeO2 from first principles, Molecular Physics, 107, pp. 443-452, 2009
- Matsui_AlCaMgOSi_1994 (Ca, Si, Mg, O, Al)
- Matsui, M., A transferable interatomic potential model for crystals and melts in the system CaO-MgO-Al2O3-SiO3, MinMag, 58, pp. 571-572, 1994
- Matsui_MgOSi_1987 (Mg, Si, O)
- Matsui, Masanori and Akaogi, Masaki and Matsumoto, Takeo, Computational model of the structural and elastic properties of the ilmenite and perovskite phases of MgSiO3, Physics and Chemistry of Minerals, 14, pp. 101-106, 1987
- Matsui_OTi_1991 (O, Ti)
- Masanori Matsui and Masaki Akaogi, Molecular Dynamics Simulation of the Structural and Physical Properties of the Four Polymorphs of TiO2, Molecular Simulation, 6, pp. 239-244, 1991
- MitchellFincham_CaF_1993 (Ca, Ca_shell, F_shell, F)
- Mitchell, P. J. and Fincham, D., Shell model simulations by adiabatic dynamics, Journal of Physics: Condensed Matter, 5, pp. 1031-1038, 1993
- MitchellFincham_MgO_1993 (Mg, O, O_shell)
- Mitchell, P. J. and Fincham, D., Shell model simulations by adiabatic dynamics, Journal of Physics: Condensed Matter, 5, pp. 1031-1038, 1993
- MitchellFincham_NaCl_1993 (Na, Na_shell, Cl_shell, Cl)
- Mitchell, P. J. and Fincham, D., Shell model simulations by adiabatic dynamics, Journal of Physics: Condensed Matter, 5, pp. 1031-1038, 1993
- Oligschleger_Se_1996 (Se)
- Oligschleger, C. and Jones, R. O. and Reimann, S. M. and Schober, H. R., Model interatomic potential for simulations in selenium, Phys. Rev. B, 53, pp. 6165-6173, 1996 link
- Pedone_2006Fe2 (Ni, Na, Nd, Li, Ti, Be, Ba, Fe, Mg, Sr, K, Mn, O, P, Si, Sn, Sc, Zn, Co, Ag, Ca, Al, Ge, Gd, Cu, Cr, Zr, Er)
- Pedone and G. Malavasi and M. Menziani and A. Cormack and U. Segre, A new self-consistent empirical interatomic potential model for oxides, silicates and silica-based glasses, J. Phys. Chem. B, 110, pp. 11780-11795, 2006
- Pedone_2006Fe3 (Ni, Na, Nd, Li, Ti, Be, Ba, Fe, Mg, Sr, K, Mn, O, P, Si, Sn, Sc, Zn, Co, Ag, Ca, Al, Ge, Gd, Cu, Cr, Zr, Er)
- Pedone and G. Malavasi and M. Menziani and A. Cormack and U. Segre, A new self-consistent empirical interatomic potential model for oxides, silicates and silica-based glasses, J. Phys. Chem. B, 110, pp. 11780-11795, 2006
- Pedone_AlCaNaOSi_2012 (Na, Ca, Al, O, Si, O_shell)
- Pedone, Alfonso and Gambuzzi, Elisa and Menziani, Maria Cristina, Unambiguous Description of the Oxygen Environment in Multicomponent Aluminosilicate Glasses from 17O Solid State NMR Computational Spectroscopy, The Journal of Physical Chemistry C, 116, pp. 14599-14609, 2012
- Pedone_LiNaKSiO_2007 (Na, K, Si, O, Li)
- Pedone, Alfonso and Malavasi, Gianluca and Cormack, Alastair N and Segre, Ulderico and Menziani, M Cristina, Insight into elastic properties of binary alkali silicate glasses; prediction and interpretation through atomistic simulation techniques, Chemistry of materials, 19, pp. 3144-3154, 2007
- Pinilla_HO_2012 (H, O)
- ReaxFF_CHF (H, C, F)
- Oliver Böhm, AQcomputare GmbH, CHF parameter set Version 4.6, 2015
- ReaxFF_CHOCaSiAlS_2012 (C, S, H, Ca, Al, O, Si)
- Liu, Lianchi and Jaramillo-Botero, Andres and Goddard III, William A and Sun, Huai, Development of a ReaxFF reactive force field for ettringite and study of its mechanical failure modes from reactive dynamics simulations, The Journal of Physical Chemistry A, 116, pp. 3918-3925, 2012
- ReaxFF_CHOFeSCr_2015 (C, H, O, S, Fe, Cr)
- Shin, Yun Kyung and Kwak, Hyunwook and Vasenkov, Alex V and Sengupta, Debasis and van Duin, Adri CT, Development of a ReaxFF reactive force field for Fe/Cr/O/S and application to oxidation of butane over a pyrite-covered Cr2O3 catalyst, ACS Catalysis, 5, pp. 7226-7236, 2015
- ReaxFF_CHOFe_2010 (H, C, Fe, O)
- Aryanpour, Masoud and van Duin, Adri C. T. and Kubicki, James D., Development of a Reactive Force Field for Iron−Oxyhydroxide Systems, The Journal of Physical Chemistry A, 114, pp. 6298-6307, 2010
- ReaxFF_CHONSFPtClNi_2010 (Ni, C, Pt, F, H, Cl, O, N, S)
- Mueller, Jonathan E. and van Duin, Adri C. T. and Goddard, William A., Development and Validation of ReaxFF Reactive Force Field for Hydrocarbon Chemistry Catalyzed by Nickel, The Journal of Physical Chemistry C, 114, pp. 4939-4949, 2010
- ReaxFF_CHONSSiCaCsKSrNaMgAlCu_2015 (C, Mg, H, Ca, Al, Na, S, O, N, Si, Sr, Cs, K, Cu)
- Psofogiannakis, George M and McCleerey, John F and Jaramillo, Eugenio and van Duin, Adri CT, ReaxFF Reactive Molecular Dynamics Simulation of the Hydration of Cu-SSZ-13 Zeolite and the Formation of Cu Dimers, The Journal of Physical Chemistry C, 119, pp. 6678-6686, 2015
- ReaxFF_CHONSSiLi_2013 (C, H, S, O, N, Si, Li)
- QuantumWise A/S, Atomistix ToolKit 2014 Reference Manual, 2014
- ReaxFF_CHONSSiPtZrNiCuCo_2005 (Ni, C, Co, Pt, H, S, O, N, Si, Cu, Zr)
- Nielson, Kevin D and van Duin, Adri CT and Oxgaard, Jonas and Deng, Wei-Qiao and Goddard, William A, Development of the ReaxFF reactive force field for describing transition metal catalyzed reactions, with application to the initial stages of the catalytic formation of carbon nanotubes, The Journal of Physical Chemistry A, 109, pp. 493-499, 2005
- ReaxFF_CHONSSi_2009 (C, H, S, O, N, Si)
- Zhang, Luzheng and Zybin, Sergey V and van Duin, Adri CT and Dasgupta, Siddharth and Goddard III, William A and Kober, Edward M, Carbon cluster formation during thermal decomposition of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine and 1, 3, 5-triamino-2, 4, 6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations, The Journal of Physical Chemistry A, 113, pp. 10619-10640, 2009
- ReaxFF_CHONSSi_2012 (C, H, S, O, N, Si)
- Kulkarni, Anant D and Truhlar, Donald G and Goverapet Srinivasan, Sriram and van Duin, Adri CT and Norman, Paul and Schwartzentruber, Thomas E, Oxygen interactions with silica surfaces: Coupled cluster and density functional investigation and the development of a new ReaxFF potential, The Journal of Physical Chemistry C, 117, pp. 258-269, 2012
- ReaxFF_CHONSSi_2012_2 (C, H, S, O, N, Si)
- Newsome, David A and Sengupta, Debasis and Foroutan, Hosein and Russo, Michael F and van Duin, Adri CT, Oxidation of Silicon Carbide by O2 and H2O: A ReaxFF Reactive Molecular Dynamics Study, Part I, The Journal of Physical Chemistry C, 116, pp. 16111-16121, 2012
- ReaxFF_CHONS_2010 (H, C, S, O, N)
- Mattsson, Thomas R. and Lane, J. Matthew D. and Cochrane, Kyle R. and Desjarlais, Michael P. and Thompson, Aidan P. and Pierce, Flint and Grest, Gary S., First-principles and classical molecular dynamics simulation of shocked polymers, Phys. Rev. B, 81, p. 054103, 2010 link
- ReaxFF_CHONSiPtZrYBaTi_2013 (C, Ba, Pt, H, O, N, Si, Ti, Y, Zr)
- Naserifar, Saber and Liu, Lianchi and Goddard III, William A and Tsotsis, Theodore T and Sahimi, Muhammad, Toward a Process-Based Molecular Model of SiC Membranes. 1. Development of a Reactive Force Field, The Journal of Physical Chemistry C, 117, pp. 3308-3319, 2013
- ReaxFF_CHONTi_2012 (H, C, N, O, Ti)
- Jaramillo-Botero, Andres and An, Qi and Cheng, Mu-Jeng and Goddard III, William A and Beegle, Luther W and Hodyss, Robert, Hypervelocity Impact Effect of Molecules from Enceladus’ Plume and Titan’s Upper Atmosphere on NASA’s Cassini Spectrometer from Reactive Dynamics Simulation, Physical review letters, 109, p. 213201, 2012
- ReaxFF_CHON_2003 (H, C, O, N)
- Strachan, Alejandro and van Duin, Adri CT and Chakraborty, Debashis and Dasgupta, Siddharth and Goddard III, William A, Shock waves in high-energy materials: The initial chemical events in nitramine RDX, Physical Review Letters, 91, p. 098301, 2003
- ReaxFF_CHON_2009 (H, C, O, N)
- Budzien, Joanne and Thompson, Aidan P and Zybin, Sergey V, Reactive molecular dynamics simulations of shock through a single crystal of pentaerythritol tetranitrate, The Journal of Physical Chemistry B, 113, pp. 13142-13151, 2009
- ReaxFF_CHONi_2015 (H, C, O, Ni)
- Tavazza, F and Senftle, TP and Zou, C and Becker, CA and van Duin, AC T, Molecular Dynamics Investigation of the Effects of Tip–Substrate Interactions during Nanoindentation, The Journal of Physical Chemistry C, 119, pp. 13580-13589, 2015
- ReaxFF_CHOSi_2005 (H, C, Si, O)
- Chenoweth, Kimberly and Cheung, Sam and Van Duin, Adri CT and Goddard, William A and Kober, Edward M, Simulations on the thermal decomposition of a poly (dimethylsiloxane) polymer using the ReaxFF reactive force field, Journal Of The American Chemical Society, 127, pp. 7192-7202, 2005
- ReaxFF_CHOV_2008 (H, C, O, V)
- Chenoweth, Kimberly and van Duin, Adri CT and Goddard, William A, ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation, The Journal of Physical Chemistry A, 112, pp. 1040-1053, 2008
- ReaxFF_CHO_2008 (H, C, O)
- Chenoweth, Kimberly and van Duin, Adri CT and Goddard, William A, ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation, The Journal of Physical Chemistry A, 112, pp. 1040-1053, 2008
- ReaxFF_C_2015 (C)
- Srinivasan, Sriram Goverapet and van Duin, Adri CT and Ganesh, P, Development of a ReaxFF potential for carbon condensed phases and its application to the thermal fragmentation of a large fullerene, The Journal of Physical Chemistry A, 119, pp. 571-580, 2015
- ReaxFF_GaN (Ga, N)
- Oliver Böhm, AQcomputare GmbH, GaN parameter set Version 4.6, 2015
- ReaxFF_HOAu_2010 (H, Au, O)
- Keith, John A. and Fantauzzi, Donato and Jacob, Timo and van Duin, Adri C. T., Reactive forcefield for simulating gold surfaces and nanoparticles, Phys. Rev. B, 81, p. 235404, 2010 link
- ReaxFF_HONB_2010 (H, B, O, N)
- Weismiller, Michael R. and Duin, Adri C. T. van and Lee, Jongguen and Yetter, Richard A., ReaxFF Reactive Force Field Development and Applications for Molecular Dynamics Simulations of Ammonia Borane Dehydrogenation and Combustion, The Journal of Physical Chemistry A, 114, pp. 5485-5492, 2010
- ReaxFF_HOSiAlLi_2012 (H, Si, Al, O, Li)
- Narayanan, Badri and van Duin, Adri CT and Kappes, Branden B and Reimanis, Ivar E and Ciobanu, Cristian V, A reactive force field for lithium–aluminum silicates with applications to eucryptite phases, Modelling and Simulation in Materials Science and Engineering, 20, p. 015002, 2012
- ReaxFF_HOZn_2010 (H, Zn, O)
- David Raymand and Adri C.T. van Duin and Daniel Spångberg and William A. Goddard III and Kersti Hermansson, Water adsorption on stepped ZnO surfaces from MD simulation, Surface Science, 604, pp. 741 – 752, 2010 link
- ReaxFF_HPd_2014 (H, Pd)
- Senftle, Thomas P and Janik, Michael J and van Duin, Adri CT, A ReaxFF investigation of hydride formation in palladium nanoclusters via Monte Carlo and molecular dynamics simulations, The Journal of Physical Chemistry C, 118, pp. 4967-4981, 2014
- ReaxFF_LiS_2015 (S, Li)
- Islam, Md Mahbubul and Ostadhossein, Alireza and Borodin, Oleg and Yeates, A Todd and Tipton, William W and Hennig, Richard G and Kumar, Nitin and van Duin, Adri CT, ReaxFF molecular dynamics simulations on lithiated sulfur cathode materials, Physical Chemistry Chemical Physics, 17, pp. 3383-3393, 2015
- ReaxFF_SiCNH (H, Si, C, N)
- Oliver Böhm, AQcomputare GmbH, SiCNH parameter set, 2015
- ReaxP_AlHOSi_2016 (H, Si, Al, O)
- B{ö}hm, Oliver and Pfadenhauer, Stephan and Leitsmann, Roman and Plänitz, Philipp and Schreiner, Eduard and Schreiber, Michael, ReaxFF+-a New Reactive Force Field Method for the Accurate Description of Ionic Systems and Its Application to the Hydrolysation of Aluminosilicates
- Rohl_OZn_1996 (Zn, Zn_shell, O, O_shell)
- Nyberg, Mats and Nygren, Martin A and Pettersson, Lars GM and Gay, David H and Rohl, Andrew L, Hydrogen dissociation on reconstructed ZnO surfaces, The Journal of Physical Chemistry, 100, pp. 9054-9063, 1996
- Schelling_OYZr_2001 (Y, Zr, O)
- Schelling, Patrick K. and Phillpot, Simon R. and Wolf, Dieter, Mechanism of the Cubic-to-Tetragonal Phase Transition in Zirconia and Yttria-Stabilized Zirconia by Molecular-Dynamics Simulation, Journal of the American Ceramic Society, 84, pp. 1609-1619, 2001 link
- StillingerWeber_BN_2005 (B, N)
- Moon, Won Ha and Hwang, Ho Jung, A modified Stillinger–Weber empirical potential for boron nitride, Applied surface science, 239, pp. 376-380, 2005
- StillingerWeber_BN_2007 (B, N)
- Moon, Won Ha and Son, Myung Sik and Hwang, Ho Jung, Theoretical study on structure of boron nitride fullerenes, Applied surface science, 253, pp. 7078-7081, 2007
- StillingerWeber_CdTeZnSeHgS_2013 (Zn, Cd, S, Te, Hg, Se)
- Zhou, XW and Ward, DK and Martin, JE and van Swol, FB and Cruz-Campa, JL and Zubia, D, Stillinger-Weber potential for the II-VI elements Zn-Cd-Hg-S-Se-Te, Physical Review B, 88, p. 085309, 2013
- StillingerWeber_MoS_2013 (Mo, S)
- Jiang, Jin-Wu and Park, Harold S and Rabczuk, Timon, Molecular dynamics simulations of single-layer molybdenum disulphide (MoS2): Stillinger-Weber parametrization, mechanical properties, and thermal conductivity, Journal of Applied Physics, 114, p. 064307, 2013
- StillingerWeber_SiGe_1995 (Si, Ge)
- Laradji, Mohamed and Landau, DP and Dünweg, B, Structural properties of Si 1-x Ge x alloys: A Monte Carlo simulation with the Stillinger-Weber potential, Physical Review B, 51, p. 4894, 1995
- StillingerWeber_SiGe_2008 (Si, Ge)
- Gabriel, Alice-Agnes, Atomistic simulation of solid-phase epitaxial regrowth of amorphous Germanium, 2008
- StillingerWeber_Si_1985 (Si)
- Stillinger and T. A. Weber, Computer simulation of local order in condensed phases of silicon, Phys. Rev. B, 31, pp. 5262-5271, 1985
- SuttonChen_Classical_1998 (Ni, Ag, Pt, Ir, Au, Pd, Rh, Cu)
- Kimura, Yoshitaka and Qi, Yue and Cagin, Tahir and Goddard, WA, The quantum Sutton–Chen many-body potential for properties of fcc metals, Phys. Rev., to be submitted, 1998
- SuttonChen_Fe_2000 (Fe)
- Belonoshko, Anatoly B and Ahuja, R and Johansson, B{ö}rje, Quasi–Ab initio molecular dynamic study of Fe melting
- SuttonChen_NiAl_2008 (Ni, Al)
- Kazanc, Sefa and Tatar, Cengiz, Investigation of the effect of pressure on some physical parameters and thermoelastic phase transformation of NiAl alloy, International Journal of Solids and Structures, 45, pp. 3282-3289, 2008
- SuttonChen_NiCuAgAuPtRh_1999 (Ni, Ag, Pt, Au, Rh, Cu)
- Cagin, T and Dereli, G and Uludogan, M and Tomak, M, Thermal and mechanical properties of some fcc transition metals, Physical Review B, 59, p. 3468, 1999
- SuttonChen_Original_1991 (Ni, Ag, Pt, Ir, Al, Pb, Au, Pd, Rh, Cu)
- Rafii-Tabar, H and Sutton, AP, Long-range Finnis-Sinclair potentials for fcc metallic alloys, Philosophical Magazine Letters, 63, pp. 217-224, 1991
- SuttonChen_Original_1998 (Ni, Ag, Pt, Ir, Au, Pd, Rh, Cu)
- Kimura, Yoshitaka and Qi, Yue and Cagin, Tahir and Goddard, WA, The quantum Sutton–Chen many-body potential for properties of fcc metals, Phys. Rev., to be submitted, 1998
- SuttonChen_Quantum_1998 (Ni, Ag, Pt, Ir, Au, Pd, Rh, Cu)
- Kimura, Yoshitaka and Qi, Yue and Cagin, Tahir and Goddard, WA, The quantum Sutton–Chen many-body potential for properties of fcc metals, Phys. Rev., to be submitted, 1998
- Tangney_OSi_2002 (Si, O)
- Tangney, Paul and Scandolo, Sandro, An ab initio parametrized interatomic force field for silica, The Journal of chemical physics, 117, pp. 8898-8904, 2002
- Tangney_OTi_2010 (O, Ti)
- Han, X. J. and Bergqvist, L. and Dederichs, P. H. and M”uller-Krumbhaar, H. and Christie, J. K. and Scandolo, S. and Tangney, P., Polarizable interatomic force field for TiO2 parametrized using density functional theory, Phys. Rev. B, 81, p. 134108, 2010 link
- Tersoff_AlGaAs_2000 (As, Al, Ga)
- Nordlund, K and Nord, J and Frantz, J and Keinonen, J, Strain-induced Kirkendall mixing at semiconductor interfaces, Computational materials science, 18, pp. 283-294, 2000
- Tersoff_AlNO_2009 (Al, O, N)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_AlNO_2009b (Al, O, N)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_Au_2012 (Au)
- Backman, M and Juslin, N and Nordlund, K, Bond order potential for gold, The European Physical Journal B, 85, pp. 1-5, 2012
- Tersoff_BNC_2000 (C, B, N)
- Matsunaga, Katsuyuki and Fisher, Craig and Matsubara, Hideaki, Tersoff potential parameters for simulating cubic boron carbonitrides, JAPANESE JOURNAL OF APPLIED PHYSICS PART 2 LETTERS, 39, pp. L48-L51, 2000
- Tersoff_BNO_2009 (B, O, N)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_BNO_2009b (B, O, N)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_BN_2003 (B, N)
- Moon, Won Ha and Son, Myung Sik and Hwang, Ho Jung, Molecular-dynamics simulation of structural properties of cubic boron nitride, Physica B: Condensed Matter, 336, pp. 329-334, 2003
- Tersoff_BeCH_2009 (H, C, Be)
- C Björkas and N Juslin and H Timko and K Vörtler and K Nordlund and K Henriksson and P Erhart, Interatomic potentials for the Be-C-H system, Journal of Physics: Condensed Matter, 21, p. 445002, 2009
- Tersoff_BeH_2009 (H, Be)
- C Björkas and N Juslin and H Timko and K Vörtler and K Nordlund and K Henriksson and P Erhart, Interatomic potentials for the Be-C-H system, Journal of Physics: Condensed Matter, 21, p. 445002, 2009
- Tersoff_BeW_2010 (Be, W)
- Bj{ö}rkas, C and Henriksson, KOE and Probst, M and Nordlund, K, A Be–W interatomic potential
- Tersoff_CH_2005 (H, C)
- Juslin, N and Erhart, P and Traskelin, P and Nord, J and Henriksson, Krister OE and Nordlund, K and Salonen, E and Albe, K, Analytical interatomic potential for modeling nonequilibrium processes in the W–C–H system, Journal of applied physics, 98, pp. 123520-123520, 2005
- Tersoff_CH_2010 (H, C)
- Juslin, N and Erhart, P and Traskelin, P and Nord, J and Henriksson, Krister OE and Nordlund, K and Salonen, E and Albe, K, Analytical interatomic potential for modeling nonequilibrium processes in the W–C–H system, Journal of applied physics, 98, pp. 123520-123520, 2005Lindsay, L and Broido, DA, Optimized Tersoff and Brenner empirical potential parameters for lattice dynamics and phonon thermal transport in carbon nanotubes and graphene, Physical Review B, 81, p. 205441, 2010
- Tersoff_C_1989 (C)
- Tersoff, Modeling solid-state chemistry: Interatomic potentials for multicomponent systems, Phys. Rev. B, 39, pp. 5566-5568, 1989
- Tersoff_C_1994 (C)
- Tersoff, Chemical order in amorphous silicon carbide, Physical Review B, 49, p. 16349, 1994
- Tersoff_C_2005 (C)
- Erhart and K. Albe, Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide, Physical Review B, 71, p. 035211, 2005
- Tersoff_C_2010 (C)
- Lindsay, L and Broido, DA, Optimized Tersoff and Brenner empirical potential parameters for lattice dynamics and phonon thermal transport in carbon nanotubes and graphene, Physical Review B, 81, p. 205441, 2010
- Tersoff_C_2012 (C)
- Bellido, Edson P. and Seminario, Jorge M., Molecular Dynamics Simulations of Ion-Bombarded Graphene, The Journal of Physical Chemistry C, 116, pp. 4044-4049, 2012
- Tersoff_ErH_2011 (H, Er)
- Peng, SM and Yang, Li and Long, XG and Shen, HH and Sun, Qing-Qiang and Zu, XT and Gao, Fei, Bond-Order Potential for Erbium-Hydride System, The Journal of Physical Chemistry C, 115, pp. 25097-25104, 2011
- Tersoff_FeC_2009 (C, Fe)
- Henriksson, Krister OE and Nordlund, K, Simulations of cementite: An analytical potential for the Fe-C system, Physical Review B, 79, p. 144107, 2009
- Tersoff_FeCu_2012 (Fe, Cu)
- Hou, Huai Yu and Wang, Rong Shan and Wang, Jing Tao and Liu, Xiang Bing and Chen, Guang and Huang, Ping, An analytic bond-order potential for the Fe–Cu system, Modelling and Simulation in Materials Science and Engineering, 20, p. 045016, 2012
- Tersoff_FePt_2007 (Fe, Pt)
- Müller and P. Erhart K. and Albe, Thermodynamics of L1-0 ordering in FePt nanoparticles studied by Monte Carlo simulations based on an analytic bond-order potential, Physical Review B, 76, p. 155412, 2007
- Tersoff_Fe_2007 (Fe)
- Müller and P. Erhart and K. Albe, Analytic bond-order potential for bcc and fcc iron—comparison with established embedded-atom method potentials, Journal of Physics: Condensed Matter, 19, p. 326220, 2007
- Tersoff_GaAs_2002 (As, Ga)
- Albe, Karsten and Nordlund, Kai and Nord, Janne and Kuronen, Antti, Modeling of compound semiconductors: Analytical bond-order potential for Ga, As, and GaAs, Physical Review B, 66, p. 035205, 2002
- Tersoff_GaAs_2008 (As, Ga)
- Hammerschmidt, Thomas and Kratzer, P and Scheffler, M, Analytic many-body potential for InAs/GaAs surfaces and nanostructures: Formation energy of InAs quantum dots, Physical Review B, 77, p. 235303, 2008
- Tersoff_GaAs_2011 (As, Ga)
- Fichthorn, Kristen A and Tiwary, Yogesh and Hammerschmidt, Thomas and Kratzer, Peter and Scheffler, Matthias, Analytic many-body potential for GaAs (001) homoepitaxy: Bulk and surface properties, Physical Review B, 83, p. 195328, 2011
- Tersoff_GaNO_2009 (Ga, O, N)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_GaNO_2009b (Ga, O, N)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_GaN_2003 (Ga, N);
- Nord and K. Albe and P. Erhart and K. Nordlund, Modelling of compound semiconductors: analytical bond-order potential for gallium, nitrogen and gallium nitride, Journal of Physics: Condensed Matter, 15, p. 5649, 2003
- Tersoff_InAs_2008 (As, In)
- Hammerschmidt, Thomas and Kratzer, P and Scheffler, M, Analytic many-body potential for InAs/GaAs surfaces and nanostructures: Formation energy of InAs quantum dots, Physical Review B, 77, p. 235303, 2008
- Tersoff_InGaAs_2000 (As, Ga, In)
- Nordlund, K and Nord, J and Frantz, J and Keinonen, J, Strain-induced Kirkendall mixing at semiconductor interfaces, Computational materials science, 18, pp. 283-294, 2000
- Tersoff_InNO_2009 (N, O, In)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_InNO_2009b (N, O, In)
- Okeke, Onyekwelu U and Lowther, JE, Molecular dynamics of binary metal nitrides and ternary oxynitrides, Physica B: Condensed Matter, 404, pp. 3577-3581, 2009
- Tersoff_O_2006 (O)
- Erhart, Paul and Juslin, Niklas and Goy, Oliver and Nordlund, Kai and Müller, Ralf and Albe, Karsten, Analytic bond-order potential for atomistic simulations of zinc oxide, Journal of Physics: Condensed Matter, 18, p. 6585, 2006
- Tersoff_Powell_2007 (Al, N, P, As, Ga, In, Sb)
- Powell, D and Migliorato, MA and Cullis, AG, Optimized Tersoff potential parameters for tetrahedrally bonded III-V semiconductors, Physical Review B, 75, p. 115202, 2007
- Tersoff_PtC_2002 (C, Pt)
- Albe, Karsten and Nordlund, Kai and Averback, Robert S, Modeling the metal-semiconductor interaction: Analytical bond-order potential for platinum-carbon, Physical Review B, 65, p. 195124, 2002
- Tersoff_Pt_2002 (Pt)
- Albe, Karsten and Nordlund, Kai and Averback, Robert S, Modeling the metal-semiconductor interaction: Analytical bond-order potential for platinum-carbon, Physical Review B, 65, p. 195124, 2002
- Tersoff_SiBN_2001 (Si, B, N)
- Matsunaga, Katsuyuki and Iwamoto, Yuji, Molecular dynamics study of atomic structure and diffusion behavior in amorphous silicon nitride containing boron, Journal of the American Ceramic Society, 84, pp. 2213-2219, 2001
- Tersoff_SiC_1989 (C, Si)
- Tersoff, Erratum: Modeling solid-state chemistry: Interatomic potentials for multicomponent systems, Physical Review B, 41, pp. 3248-3248, 1990; Tersoff, Modeling solid-state chemistry: Interatomic potentials for multicomponent systems, Phys. Rev. B, 39, pp. 5566-5568, 1989
- Tersoff_SiC_1994 (C, Si)
- Tersoff, Chemical order in amorphous silicon carbide, Physical Review B, 49, p. 16349, 1994
- Tersoff_SiC_1998 (Si, C)
- Devanathan, R and Diaz de la Rubia, T and Weber, WJ, Displacement threshold energies in beta-SiC, Journal of nuclear materials, 253, pp. 47-52, 1998
- Tersoff_SiC_2005 (C, Si)
- Erhart and K. Albe, Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide, Physical Review B, 71, p. 035211, 2005
- Tersoff_SiGeO_2013 (Si, Ge, O)
- Chuang, Claire Y and Li, Qiming and Leonhardt, Darin and Han, Sang M and Sinno, Talid, Atomistic analysis of Ge on amorphous SiO2 using an empirical interatomic potential, Surface Science, 609, pp. 221-229, 2013
- Tersoff_SiGeO_LT_2013 (Si, Ge, O)
- Chuang, Claire Y and Li, Qiming and Leonhardt, Darin and Han, Sang M and Sinno, Talid, Atomistic analysis of Ge on amorphous SiO2 using an empirical interatomic potential, Surface Science, 609, pp. 221-229, 2013
- Tersoff_SiGe_1989 (Si, Ge)
- Tersoff, Erratum: Modeling solid-state chemistry: Interatomic potentials for multicomponent systems, Physical Review B, 41, pp. 3248-3248, 1990; Tersoff, Modeling solid-state chemistry: Interatomic potentials for multicomponent systems, Phys. Rev. B, 39, pp. 5566-5568, 1989
- Tersoff_SiNH_1999 (H, Si, N)
- de Brito Mota, F and Justo, JF and Fazzio, A, Hydrogen role on the properties of amorphous silicon nitride, Journal of applied physics, 86, pp. 1843-1847, 1999
- Tersoff_SiO_2007 (Si, O)
- Munetoh and T. Motooka and K. Moriguchi and A. Shintani, Interatomic potential for Si–O systems using Tersoff parameterization, Computational materials science, 39, pp. 334-339, 2007
- Tersoff_Si_1988 (Si)
- Tersoff, New empirical approach for the structure and energy of covalent systems, Physical Review B, 37, p. 6991, 1988;Tersoff, Empirical interatomic potential for silicon with improved elastic properties, Physical Review B, 38, pp. 9902-9905, 1988
- Tersoff_Si_1988b (Si)
- Tersoff, Empirical interatomic potential for silicon with improved elastic properties, Physical Review B, 38, pp. 9902-9905, 1988
- Tersoff_Si_2005 (Si)
- Erhart and K. Albe, Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide, Physical Review B, 71, p. 035211, 2005
- Tersoff_WCH_2005 (H, C, W)
- Juslin, N and Erhart, P and Traskelin, P and Nord, J and Henriksson, Krister OE and Nordlund, K and Salonen, E and Albe, K, Analytical interatomic potential for modeling nonequilibrium processes in the W–C–H system, Journal of applied physics, 98, pp. 123520-123520, 2005; Erhart and K. Albe, Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide, Physical Review B, 71, p. 035211, 2005
- Tersoff_WCH_2005b (H, C, W)
- Juslin, N and Erhart, P and Traskelin, P and Nord, J and Henriksson, Krister OE and Nordlund, K and Salonen, E and Albe, K, Analytical interatomic potential for modeling nonequilibrium processes in the W–C–H system, Journal of applied physics, 98, pp. 123520-123520, 2005; Erhart and K. Albe, Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide, Physical Review B, 71, p. 035211, 2005
- Tersoff_WH_2011 (H, W)
- Li, Xiao-Chun and Shu, Xiaolin and Liu, Yi-Nan and Gao, Fei and Lu, Guang-Hong, Modified analytical interatomic potential for a W–H system with defects, Journal of Nuclear Materials, 408, pp. 12-17, 2011
- Tersoff_ZnO_2006 (Zn, O)
- Erhart, Paul and Juslin, Niklas and Goy, Oliver and Nordlund, Kai and Müller, Ralf and Albe, Karsten, Analytic bond-order potential for atomistic simulations of zinc oxide, Journal of Physics: Condensed Matter, 18, p. 6585, 2006
- Tersoff_Zn_2006 (Zn)
- Erhart, Paul and Juslin, Niklas and Goy, Oliver and Nordlund, Kai and Müller, Ralf and Albe, Karsten, Analytic bond-order potential for atomistic simulations of zinc oxide, Journal of Physics: Condensed Matter, 18, p. 6585, 2006
- Trinastic_HfOSiTaTi_2013 (Si, Ta, Hf, O, Ti)
- Trinastic, JP and Hamdan, R and Wu, Y and Zhang, L and Cheng, Hai-Ping, Unified interatomic potential and energy barrier distributions for amorphous oxides, The Journal of chemical physics, 139, p. 154506, 2013
- VanBeest_SiOAlP_1990 (P, Si, Al, O)
- Van Beest, BWH and Kramer, GJ and Van Santen, RA, Force fields for silicas and aluminophosphates based on ab initio calculations, Physical Review Letters, 64, p. 1955, 1990
- Wang_HfOZr_2012 (Hf, O, Zr)
- Wang, Yin and Zahid, Ferdows and Wang, Jian and Guo, Hong, Structure and dielectric properties of amorphous high-kappa oxides: HfO2, ZrO2, and their alloys, Phys. Rev. B, 85, p. 224110, 2012 link
- Yasukawa_HOSi_1996 (H, Si, O)
- Yasukawa, Akio, Using An Extended Tersoff Interatomic Potential to Analyze The Static-Fatigue Strength of SiO2 under Atmospheric Influence, JSME international journal. Ser. A, Mechanics and material engineering, 39, pp. 313-320, 1996
- Yasukawa_OSi_2003 (Si, O)
- Yasukawa, Akio, An Interatomic Potential for Strength Analysis under Atomospheric Influence, Ibaraki district conference, 2003, pp. 71-72, 2003
ASAP potential parameter sets
- BrennerCalculator(H, C, Si, Ge)
- D.W. Brenner and O. A. Shenderova and J. A. Harrison and S. J. Stuart and B. Ni and S. B. Sinnott, A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons, J. of Phys.: Condens. Matter, pp. 783-802, 2002.
- EMTCalculator(Ni, Cu, Pd, Ag, Pt, Au, Al)
- Jacobsen, K.W. and Stoltze, P. and Nørskov, J.K., A semi-empirical effective medium theory for metals and alloys, Surface Science, 366, pp. 394-402, 1996.