参考文献: B. Joo and E.-G. Kim, “Model-Independent Determination of the Degree of Charge Transfer in Molecular and Metal Complexes,” Chem. Commun. 51, 15071–15074 (2015). 电荷转移在化学各个领域都普遍存在,理论化学家们对此已经有了大量的“例行”研究。但这样的研究中,对原子电荷的定义是有问题的。拿Mulliken电荷来说,大家对它又爱又恨。爱,是因为用起来很简单;恨,是因为有时候会得到惊人的错误结果,并且与深受使用基组的影响。Mulliken电荷估计Ir(ppy)3的电荷转移几乎为0;对金属配体电荷转移(MLCT)标志性材料OLED得到完全不符合物理预期的结果。 本文发现了一种解决这个长期悬而未决的问题的办法(实际上并没有真正引进任何新的理论):跟踪电子转移过程中的最弥散电子,这种方法完全于具体模型无关。虽然他们的方法在技术上违背直观,因为只关注弥散电子,但物理上比其他划分方法更合理,因为只追踪参与电荷转移的电子。 这种方法描述的电荷转移的定量结果与测量结果、化学直觉完全一致。他们的方法能够扩展到其他与基态、激发态存在分子内、分子间电荷转移相关的化学领域。 关键词:charge transfer; atomic charges; bonding analysis

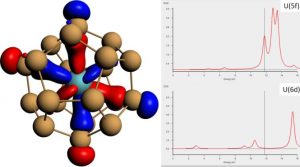
ADF Highlight:锕系原子在分子笼中的体系满足32电子原则(Chem. Sci.,2012)
参考文献: J.-P. Dognon, C. Clavaguéra and P. Pyykkö, A new, centered 32-electron system: the predicted [U@Si20]6–like isoelectronic series Chem. Sci., 3, 2843 (2012) 本文预测了一种化合物满足32电子原则。被 Chemical Science和Nature Chemistry作为亮点文章。研究了[U@Si20]6- 的性质以及等电子post-U同类化合物。这种化合物典型地填充了完整的7s,7p,6d和5f壳层,总共32个电子。Pu@Pb12和U@C28 也是如此,在早期的工作中已经作为亮点发表。成键的细节使用各种工具,包括EDA、AIM、pDOS进行了研究。 关键词:chemical analysis, TDDFT, ZORA
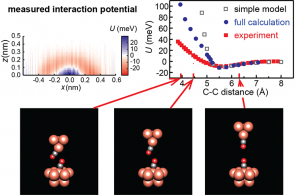
ADF Highlight:CO修饰针尖定量原子力显微镜提高分辨率(PRL,2011)
参考文献: Z. Sun, M. P. Boneschanscher1, I. Swart, D. Vanmaekelbergh, and P. Liljeroth,Quantitative Atomic Force Microscopy with Carbon Monoxide Terminated Tips. Physical Review Letters 106, 046104 (2011). 原子力显微镜(AFM)通过针尖吸附一个一氧化碳分子提高对机分子成像分辨率,本文详细研究了这种成像机制,以及分子弛豫在力谱实验中的角色。 该实验涉及两个CO分子间力的测量手段:一个CO分子吸附在Cu(111)单晶基底上,另一个在AFM针尖顶部。实验结果与ADF计算结果进行了比较。使用TZ2P基组,色散修正泛函PBE-D考虑范德华力,范德华力决定CO-CO分子之间的距离。 本文在两个层次上进行了计算:1)半定量层次,简单计算两个孤立CO分子之间的相互作用,来重现实验结果,但针尖-样品距离很短的时候,排斥作用被强烈过大估计;2)使用Cu4和Cu10团簇分别代表针尖模型及基底,包括了CO分子吸附结构的弛豫。 针尖-基底距离较小的时候,Pauli排斥力很强,会导致吸附的CO分子方向改变,来降低排斥力。这使得计算得到的相互作用能和实验更接近。 近距离中原子级别的重构限制了成像和力谱分辨率。这种效应在所有的用到分子针尖的非接触AFM实验中很普遍。 关键词:dispersion-corrected DFT (DFT-D)
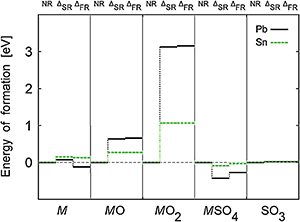
ADF Highlight:相对论效应提高电池电压
参考文献: R. Ahuja1, A. Blomqvist, P. Larsson, P. Pyykkö, and P. Zaleski-Ejgierd,Relativity and the Lead-Acid Battery. Physical Review Letters 106, 018301 (2011). 传统的铅-氧化铅电池已经有一个世纪的历史了,能够有实际用途,是由于每个伏打电池单元能够产生2V以上的电压,多个电池串联能够达到驱动电动机,锡化学性质与铅非常相近,但与铅相比,相对论效应小很多,导致锡电池产生的电压也远小于铅。 本文从理论上阐明了铅、锡以及去除相对论效应的假想铅的巨大差别。 该计算使用ADF中的BAND模块进行计算。使用了ZORA方法计算相对论效应。 关键词:periodic DFT with BAND, relativistic effects (scalar and spin-orbit coupling) with ZORA

ADF Highlight:TiO2纳米晶体定向的聚集(Nano Lett.,2014)
参考文献: M. Raju, A. C. T. van Duin, and K. A. Fichthorn, Mechanisms of Oriented Attachment of TiO2 Nanocrystals in Vacuum and Humid Environments: Reactive Molecular Dynamics, Nano Lett. 14, 1836-1842 (2014). 本文使用ReaxFF研究TiO2纳米颗粒生长,在真空中,颗粒聚集具有取向性;在水相中纳米晶体在聚集之前有时间重新定向,从而生长出单晶或孪晶。定向聚集机制主要发生强烈地奋力水分子的表面。这代表了溶剂调制晶体生长的效果。 水相中纳米颗粒能够连接、脱离、转动、找到最佳结合方式。图中左边的两个颗粒需要25ps才能调整到锐钛矿的(112)方向结合。 关键词:ReaxFF, molecular dynamics, crystal growth
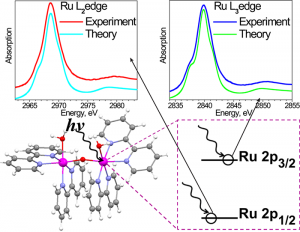
ADF Highlight:双钌催化剂的钌L2,3边沿的XANES谱(JACS,2011)
参考文献: I. Alperovich, G. Smolentsev, D. Moonshiram, J.W. Jurss, J.J. Concepcion, T.J. Meyer, A. Soldatov, Y. Pushkar Understanding the Electronic Structure of 4d Metal Complexes: From Molecular Spinors to L-Edge Spectra of a di-Ru Catalyst J. Am. Chem. Soc., 133 15786-15794 (2011). L2,3边沿的x射线吸收近边结构(XANES)谱,从2p到εs和εd能级的跃迁,包含材料和分子中过渡金属电子结构的重要信息。正确理解XANES谱需要非常熟练的计算能力。影响3d的x射线谱的因素有几种,4d、5d则不太受影响。但考虑自旋-轨道耦合非常关键,因为一些从2p1/2、2p3/2简并态到εd的跃迁是对称性禁阻的。 本文使用ADF2010计算了分子的电子自旋轨道的空间分布和钌2p能级到空轨道跃迁的XANES谱。 关键词:X-ray Absorption Near-Edge Spectroscopy, core excitations, spin-orbit coupling, TDDFT

ADF Highlight:模拟种元素化合物的32电子规则(JACS,2009; Angew. Chem. Int. Ed.,2007)
参考文献: J.-P. Dognon, C. Clavagura and P. Pyykkö, A Predicted Organometallic Series Following a 32-Electron Principle: An@C28 (An = Th, Pa+, U2+, Pu4+).Journal of the American Chemical Society, 131 (1), 238 (2009). J.-P. Dognon, C. Clavagura and P. Pyykkö, Towards a 32-Electron Principle: Pu@ Pb12 and Related Systems. Angewandte Chemie International Edition,46 (9), 1427 (2007). […]
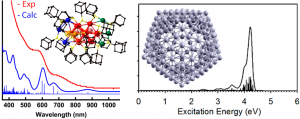
ADF Highlight:银与金纳米颗粒的光学性质(JACS,2014)
参考文献: D. Crasto, G. Barcaro, M. Stener, L. Sementa, A. Fortunelli, and A. Dass, Au24(SAdm)16 Nanomolecules: X‑ray Crystal Structure, Theoretical Analysis, Adaptability of Adamantane Ligands to Form Au23(SAdm)16 and Au25(SAdm)16, and Its Relation to Au25(SR)18, J. Am. Chem. Soc., 136, 14933-14940 (2014) G. Barcaro, L. Sementa, A. Fortunelli, and M. Stener, Optical Properties of Silver Nanoshells from Time-Dependent Density Functional Theory […]
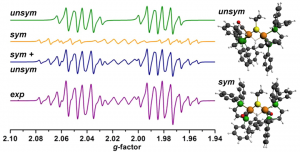
ADF Highlight:使用EPR表征析氢反应催化剂(JACS,2013)
参考文献: W. Wang, M. J. Nilges, T. B. Rauchfuss, and M. Stein, Isolation of a Mixed Valence Diiron Hydride: Evidence for a Spectator Hydride in Hydrogen Evolution Catalysis J. Am. Chem. Soc. 135, 3633-3639 (2013) 由于进一步发展氢经济的需求,双铁化合物,仿[FeFe]-H2已经作为更廉价、更具可持续性的HER催化剂被广泛研究。University of Illinois and the MPI for Dynamics of Complex Technical Systems的研究者分离出并表征了混合价态双铁氢。为仿生HER催化领域展开新的视角。 本文使用ADF的ZORA-DFT计算得到的几何结构、IR位移、EPR参数与实验结果非常相符,从而洞悉对不对称和对称异构体的自旋分布。 关键词:EPR, IR, ZORA, spin distribution
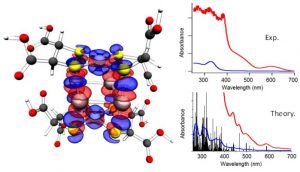
ADF Highlight:双金属纳米团簇的原子精度表征(JACS,2013)
参考文献: S. R. Biltek, S. Mandal, A. Sen, A. C. Reber, A. F. Pedicini, S. V. Khanna,Synthesis and Structural Characterization of an Atom-Precise Bimetallic Nanocluster, Ag4Ni2(DMSA)4 , J. Am. Chem. Soc. 135, 26-29 (2013) 本文中,首次合成了贵金属和第一排过渡金属元素的双金属纳米团簇,并使用实验手段(ESI-MS)和DFT理论计算结合,进行表征。 DFT计算确认出这种复杂纳米颗粒最稳定的结构——Ag4垂直平面、Ni顶结构。使用ADF的片段分析功能揭示了纳米颗粒的超强稳定性是由于银原子到镍原子之间的电荷转移引起的。结构的空间优势使得Ag-S键的数目最大化。Ag-Ni团簇的UV/VIS计算结果与实验结果极其相符。 左图是Ag4Ni2(DMSA)4的结构和片段分析,红色表示电荷密度的降低区域,蓝色是电荷密度增强的区域。右图是实验与计算得到的双金属团簇的UV/VIS光谱。 关键词:chemical analysis, TDDFT, ZORA ADF相关功能中文手册: 如何进行简单的片段分析、键能、键解离能、结合能计算 ETS-NOCV计算:以H-CN为例,拆分为H、CN中性片段 ETS-NOCV计算:以H-CN为例,拆分为H+、CN-带电片段 ETS-NOCV计算:以环己烯为例,拆分为两个三重态的片段 文献重现:使用ETS-NOCV自然价键轨道研究化学反应 文献重现:氢键强度、轨道作用、电荷分析 如何进行开壳层片段的分析