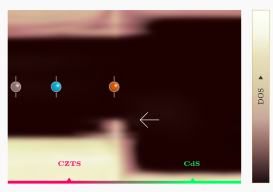
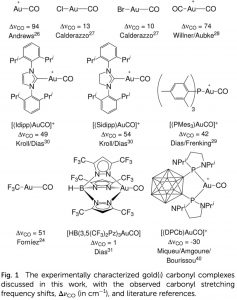
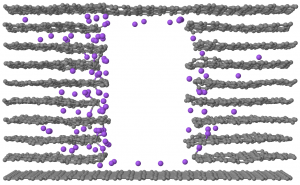
在Applied Physics Letters 2017 年发表的最新文章中使用 QuantumATK 讨论了在 Cu2ZnSnS4 太阳能电池体系里材料界面处能带收窄导致的开路电压损失。文章由 Synopsys QuantumWise 公司和丹麦技术大学(DTU)的研究者共同完成。 Cu2ZnSnS4(CZTS)是很有前景的薄膜光电功能材料。由于其具有直接带隙,因此 CZTS 构成的器件可以比间接带隙的硅基器件更有效的吸收光。此外,CZTS 体系中只包含地球上丰度很高的元素,因此比诸如 CdTe 等体系更具有经济价值。 目前 CZTS 体系主要的问题是开路电压(OCV)很低,并且原因不明。目前主流的解释是 CZTS 与作为缓冲层的 CdS 之间的(大台阶状)能带对齐是主要的不利因素,然而,目前对最新的效率超过7%的CZTS器件的测量都表明能带对齐相当的合适(平坦)。 本论文从另外的角度来解释 OCV 损失。研究者使用 QuantumATK 计算了 CZTS 和 CdS 之间界面处的电子态(能带对齐)情况,并在界面处发现了一处很小的局域化的界面态。在进行器件级别模拟时,此界面态导致了显著的 OCV 衰减,本文给出的计算结果与文献中报道的目前 CZTS 光电池器件测量结果定量的吻合。本研究显示了器件级别模拟所需参数可以从原子级别的材料模拟结果中获得,将两个级别的模拟结合可以为未来薄膜光电池器件提供有力的工具。 QuantumATK 中提供: 便捷的材料界面建模工具,构建任意表面之间的界面模型; 使用双电极器件模型(device model)与 NEGF 方法直接计算能带对齐,避免 Slab 模型能级对齐的困扰。 参考文献 文章原文:A. Crovetto, M. Palsgaard, T. Gunst, […]