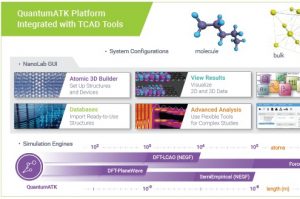
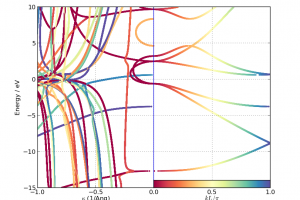
新一代材料与器件模拟平台QuantumATK的“终极”参考文献“QuantumATK: an integrated platform of electronic and atomic-scale modelling tools”已在线发布![1] 除了提供总体概述和一些以前未发布的方法实现细节外,文章还提供了四个重要的应用示例: Cu、Ag和Au的声子限制迁移率 门控二维器件中的电子输运 锂离子在外电场中通过电池正极漂移的多模型模拟 SiGe合金成分相关带隙的电子结构计算 要了解更多新一代材料与器件模拟平台的概况和使用方法,敬请关注《材料学计算模拟系列课程:QuantumATK材料与器件模拟平台使用入门(11月23-24日,西安)》。 如何正确引用QuantumATK 从现在起,请 QuantumATK 的用户在任何发表使用软件得到的结果刊物中引用本文。我们建议您以以下格式提供所用软件的版本: “QuantumATK: An integrated platform of electronic and atomic-scale modelling tools”, S. Smidstrup et al., J. Phys.: Condens. Matter 32, 015901 (2020). QuantumATK, version P-2019.03, https://www.synopsys.com/silicon/quantumatk.html QuantumATK材料模拟平台概述 模拟引擎 QuantumATK模拟引擎可以使用密度泛函理论(DFT)或紧束缚模型哈密顿量进行电子结构计算,还可以在许多不同参数下提供键合或反应型经验力场。DFT可以在平面波基组或原子轨道线性组合(LCAO)基组展开电子态来实现计算。 图形用户界面 NanoLab:基于插件的图形用户界面(GUI),所有QuantumATK模拟引擎都集成于统一的GUI NanoLab links:使NanoLab能够连接其他代码的功能模块 插件服务器:为NanoLab下载数百个不同的专业功能模块 原子结构 分子(非周期体系) […]