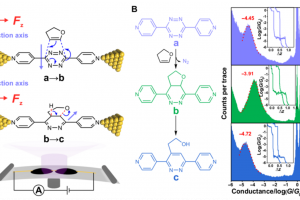
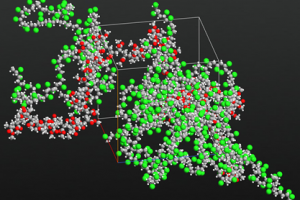
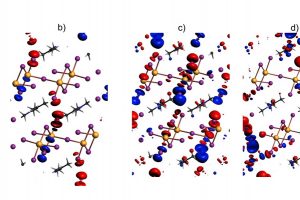
引言 随着单分子电学检测技术的迅速发展,分子电子学的研究已不再局限于分子电子学器件的构筑及其电学性质的测量,而是扩展到单分子尺度化学反应过程的探索。单分子裂结技术能够直接观察单个分子的始态、终态和中间态,采用此技术并通过施加定向外部电场,有望实现对单个分子乃至单个化学反应的操纵和调控,从而在纳米尺度的反应器中实现对单个分子化学反应速率的选择性调控,为未来基于清洁能源的绿色化学合成提供了新思路。近日,厦门大学洪文晶教授、程俊教授和兰州大学张浩力教授在电场选择性调控特定化学键的反应速率研究方面取得重要突破。相关研究成果以“Electric-field-induced selective catalysis of single-molecule reaction”为题发表于Science Advances。 成果简介 针对这一挑战,该研究团队自主研发了高强度定向电场下研究化学反应速率的精密科学仪器技术,将单个有机分子定向地连接在两个原子级尺寸的电极之间,从而解决了化学反应中分子取向控制的问题。通过电极对单个分子施加了高达108-9 V/m的定向电场和对反应的分子计数,精确测量了两步连续反应中每步的反应速率。在纳米尺度反应器内中发现:若施加电场与反应轴垂直,电场对化学反应没有影响;如果电场在反应轴方向有分量,电场可以使反应速率提升超过一个数量级。通过单分子器件电输运模拟证实了化学反应路径的中间态;过渡态计算结果表明定向电场可以有效地稳定化学反应的过渡态,从而降低反应能垒。 这一跨学科合作,在国际上首创了利用定向外部电场选择性调控化学反应速率的仪器和技术,实现外加电场对化学反应的有效调控,提出了将外加电场作为智能催化剂创新思路,对基于清洁能源的绿色化学合成和化工清洁生产的发展具有重要意义。 参考文献 X. Huang, C. Tang, J. Li, L. C. Chen, J. Zheng, P. Zhang, J. Le, R. Li, X. Li, J. Liu, Y. Yang, J. Shi, Z. Chen, M. Bai, H. L. Zhang, H. Xia, J. Cheng, Z. Q. Tian and W. Hong. […]