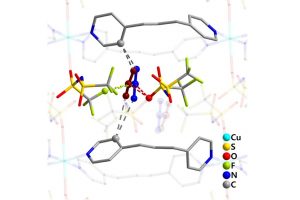
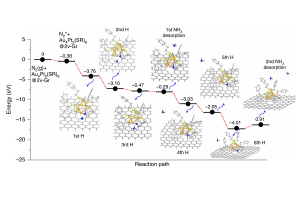
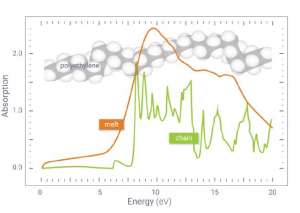
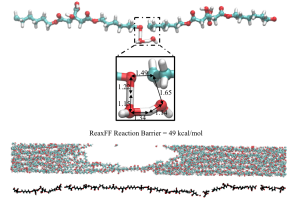
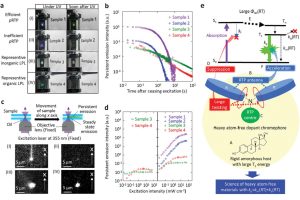
离子液体(ILs)的二氧化碳(CO2)选择性吸收特性与CO2捕集方法的发展密切相关。尽管有报道称氟化组分使ILs增强了CO2溶解度,但深入理解ILs与CO2之间的相互作用一直是一个挑战。在本研究中,作者利用软晶质材料[Cu(NTf2)2(bpp)2] (NTf2‒ = bis(trifluoromethylsulfonyl)imide, bpp = 1,3-bis-(4-pyridyl)propane)作为单晶X射线衍射分析的替代物,将CO2与NTf2‒(氟化离子液体组分,导致二氧化碳高溶解度)之间的相互作用可视化。对负载二氧化碳的晶体结构的分析表明,CO2与NTf2‒阴离子的氟原子和氧原子以反式而非顺式结构发生相互作用。对负载CO2的晶体结构的理论分析表明,CO2与骨架之间存在色散和静电相互作用。总而言之,为理解和改进离子液体吸收二氧化碳的特性提供了重要的见解。 使用AMS-ADF优化添加H原子的结构(PBE-D3(BJ)/TZ2P),并使用能量分解方法(EDA)结合化学价态理论的自然轨道对模型(NOCV)结构进行了分析。 参考文献: Xin Zheng, Katsuo Fukuhara, Yuh Hijikata, Jenny Pirillo, Hiroyasu Sato, Kiyonori Takahashi, Shin-ichiro Noro & Takayoshi Nakamura, Understanding the interactions between the bis(trifluoromethylsulfonyl)imide anion and absorbed CO2 using X-ray diffraction analysis of a soft crystal surrogate, Communications Chemistry volume 3, Article number: 143 (2020)