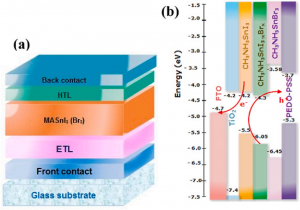
概述 此项研究使用两种计算方法来研究混合钙钛矿型锡基光伏太阳能电池。第一种方法基于微观的原子级别电子输运性质计算,结合了密度泛函理论和非平衡格林函数理论。作者对透射谱和态密度的模拟结果表明,由于电子态的离域化,从 MASnI3 到 MASnBr3 的输运带隙减小,表现出较大的电子输运能力。第二种方法是基于器件尺度的漂移扩散方法的模拟,发现钙钛矿-锡基混合光伏电池的参数明显依赖于钙钛矿吸收层的厚度。表面复合速度在 1~10 cm/s 和 102~103 cm/s 范围内,MASnIBr2 和 MASnBr3 的效率分别达到 16.07% 和 12.52%,可以提高太阳能电池的性能。 研究中使用 QuantumATK 直接构造了如下的器件模型,这在QuantumATK的图形用户界面中可以十分方便的快速完成。 研究中采用了QuantumATK中最为成熟的DFT-NEGF方法计算了上述系列器件的伏安特性、能带排列等性质。 作者还采用免费软件 SCAPS-1D 对真实尺寸的器件性能进行了研究,比如性能对吸光层厚度的依赖等。在对多层体系的能带排列进行研究时,QuantumATK提供了更方便的工具,并可以将结果与器件尺度的研究耦合起来: 双端电极界面模型可以免除Slab模型的偶极校正等烦恼 PLDOS计算模块直接得到多层体系的能带排列 快速的杂化泛函方法(HSE06-LCAO)研究更大的体系 参考 光伏材料的计算模拟与器件仿真 使用QuantumATK进行多尺度模拟:界面处能带收窄导致CZTS太阳能电池开路电压损失 材料界面的建模和模拟 原文:Physica B: Physics of Condensed Matter 591 (2020) 412247. 立即试用 QuantumATK! 下载QuantumATK软件安装包 申请QuantumATK的全功能试用许可