概述
随着计算机性能与计算理论方法的改进,在原子分子水平模拟分子的结构与行为,在石油化工领域得到越来越广泛的应用。在高分子材料、分子筛催化剂以及油品添加剂的研发中,计算模拟能够帮助研究者更深入地理解所研究的体系,促进新分子筛催化剂、高分子材料的的开发与改性,油品添加剂新产品研制,减少实验工作,缩短研发周期。
分子筛催化
- 结构
- 分子筛存在大量的真空区域,基于平面波的DFT计算将耗费大量时间在无意义的真空区域。BAND采用STO+NO基组,从而大大节省计算时间,使用GGA等泛函进行结构优化具有较好的可行性
- 吸附与扩散
- BAND、DFTB提供D3(BJ)、D4(EEQ)色散修正,更精确的计算分子吸附
- 自动探索表面吸附位点,自动探索表面化学反应机理
- 通过基于DFT、MOPAC、DFTB或力场的分子动力学模拟能够非常便捷地计算扩散系数
- 分子筛与分子之间相互作用能量的分解分析,以及电子转移的定性图像
- 巨正则系综蒙特卡洛、分子动力学模拟MOF材料表面的吸附等温线
高分子催化
- 高分子聚合催化剂的结构优化
- 高效并行的ADF能够支持大分子的DFT结构优化
- 支持多尺度方法,用户可以灵活划定聚合物原子区域,对不同区域采用不同的理论,协同优化聚合催化剂结构
- 对于一维周期性结构BAND采用的STO+NO基组大大提高计算效率,能够以比相对于平面波方法低一个数量级以上的计算成本,完成高精度GGA结构优化
- 高分子催化反应机理
- 吸附结构优化
- 多尺度方法计算反应过渡态、活化能、反应历程
- 对于一维周期性结构,支持高精度、高效率的DFT过渡态搜索、活化能计算、反应历程
- 使用ReaxFF分子动力学模拟对聚合物的力学性质,如杨氏模量、屈服点、泊松比进行预测
- ReaxFF分子动力学探索未知反应机理
- 根据反应物、产物的分子结构,筛选反应通道
- 快速确认(副)反应可能性、(副)产物可能性
- 确定化学反应中反应物和产物之间的最佳原子映射
- 聚合物与分子之间相互作用能量的分解分析,以及电子转移的定性图像
油品添加剂
- 热解与燃烧
- DFTB、MOPAC、ReaxFF均可作为分子动力学模拟引擎,支持同样的结果分析工具;ReaxFF作为经典的热解与燃烧分子模拟工具,包含最丰富的反应力场,广泛应用于该领域的研究
- Bond Boost、REMD、CVHD、fbMC等加速反应方法,能够让分子动力学模拟在实
- 验温度下,得到宏观时间尺度化学反应的结果
- Mol Sink允许在分子动力学模拟过程中,定时清除指定产物;Mol Gun可以定时添加反应物
- 自动分析产物数量变化、基元反应、反应速率常数,评估

其他研究工具
- COSMO-RS
- 气-液相平衡、液-液相平衡
- 最优萃取溶剂优化
- 活度系数、溶解度、pKa、logP
- 使用定量构效关系估算物质性质:密度、熔点、沸点、闪点、介电常数、液态摩尔体积、分子范德华体积与表面积等
- EDA-NOCV
- 键能分解分析,对分子间范德华作用、氢键作用能进行分解分析,分析泡利排斥、静电作用、轨道作用、色散作用能大小
- 分析化学键形成机理,电子在分子轨道中的定量转移,以及定性图像
- 固体表面与分子之间相互作用能量的分解分析,以及电子转移的定性图像
- 分子动力学
- 粘度
研究实例
实例1:Co/Mn/Na/S催化高选择性费托反应
Jingxiu Xie, Pasi P. Paalanen, Tom W. van Deelen, Bert M. Weckhuysen, Manuel J. Louwerse & Krijn P. de Jong
Promoted cobalt metal catalysts suitable for the production of lower olefins from natural gas
Nature Communications, volume 10, Article number: 167 (2019)
近年来,由于天然气产量激增,化学原料需求向较轻的碳氢化合物,特别是甲烷转移。
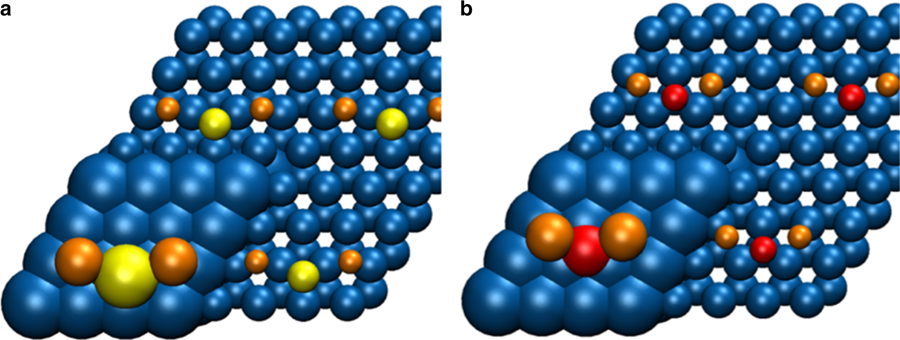
本文作者设计了一种Co/Mn/Na/S催化剂,该催化剂的水-气转换活性可以忽略不计,烃的产物谱也偏离了Anderson-Schulz-Flory分布。在240 °C和1 bar压强下,C2-C4烯烃选择性为54%。10 bar压强下,低碳烯烃和燃料的选择性分别为30%和59%。该催化剂由直径约10 nm的处于hcp金属相Co纳米颗粒组成。作者认为Na与S在Co表面作为电子促进剂,产生协同效应,从而提高了对低碳烯烃与燃料的选择性,同时也大大减少了甲烷和二氧化碳的生成。
文中使用AMS软件中的BAND模块计算了Na2S和Na2O在Co(0001)表面的成键结构。
实例2:费-托催化中的助催化剂
J. Xie, J. Yang, A.I. Dugulan, A. Holmen, D. Chen, K.P. de Jong, and M.J. Louwerse
Size and Promoter Effects in Supported Iron Fischer-Tropsch Catalysts: Insights from Experiment and Theory
ACS Catal. 6, 3147-3157 (2016)
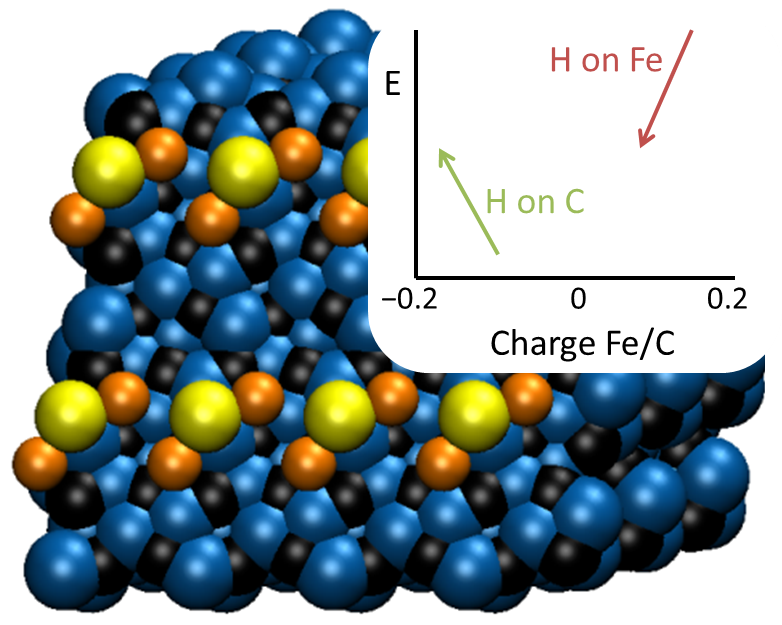
费-托反应主要目的是使用合成气生成C2到C4烯烃,该反应中,通常会产生大量不希望得到的甲烷。Utrecht的计算化学家使用AMS中BAND模块进行理论计算结合实验,合理解释了铁基费-托催化中的助催化剂效果。助催化剂的加入改进了选择性,尤其是纳与硫是一个非常好的组合。
AMS-BAND的高质量计算解释了这种特别的助催化剂组合:电荷贡献配合特殊结构达到了这种效果。催化剂的活性相是铁的碳化物,其中碳参与了反应机理。因为硫原子与铁原子的结合很特别,不会阻挡碳原子,产生了次紧邻相互作用,从而将助催化剂的作用最大化。纳硫是铁基费-托催化的很好的助催化剂
BAND模块能够正确地处理表面的边界条件,因此能够体现助催化剂和/或衬底在垂直于表面的极化效应。表面的计划对催化效果有很大的影响,因为电荷的分布能够左右反应路径。
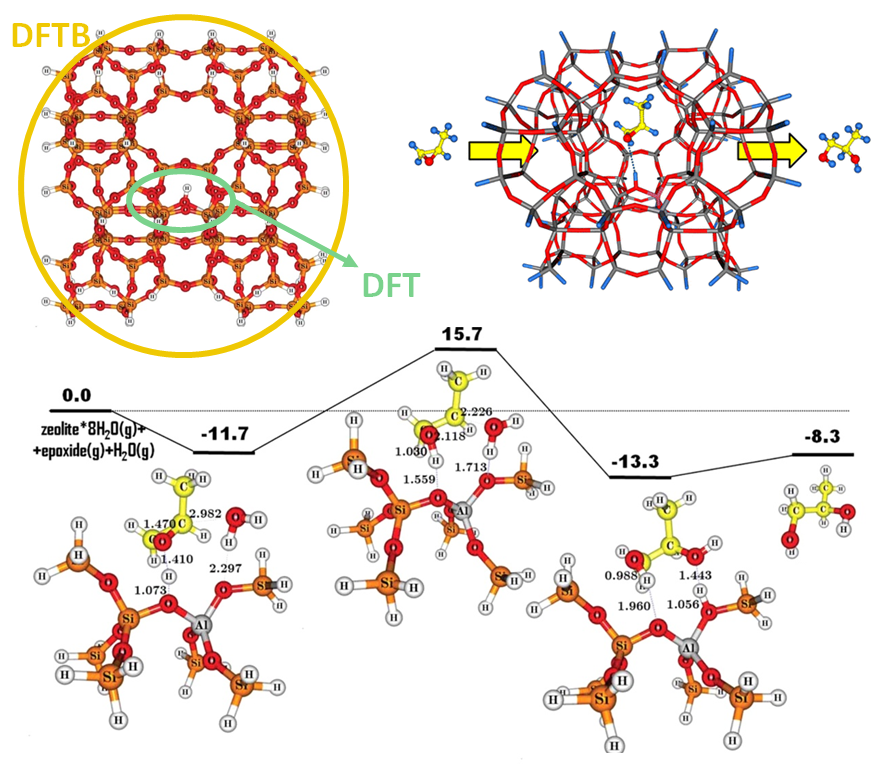
实例3:ZSM-5分子筛上环氧丙烷水解制备单丙二醇和二丙二醇的反应机理
Y. Horbatenko, J. P. Pérez, P. Hernández, M. Swart, and M. Solà
Reaction Mechanisms for the Formation of Mono- And Dipropylene Glycol from the Propylene Oxide Hydrolysis over ZSM-5 Zeolite
J. Phys. Chem. C, 118, 21952–21962 (2014)
本文使用ADF的分层(DFT/DFTB)计算功能QUILD,研究了环氧丙烷在ZSM-5中的催化水解。在精确计算的“层”使用DFT的BP86-D3泛函,在较低精度的“层”使用DFTB3计算。Monopropylene Glycol与Dipropylene Glycol通过一种协同机制优先形成,其中前者的形成能垒较低。减小孔的尺寸,可以进一步增加选择性,减弱后者的形成。