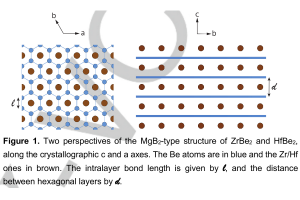
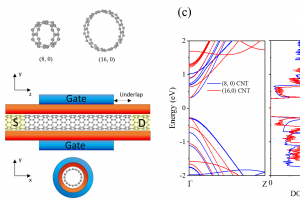
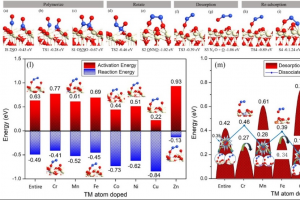
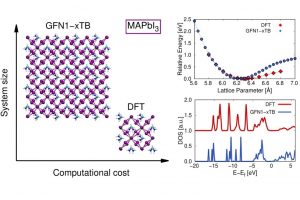
实例1:铋基半导体材料作为光催化剂 近年来,铋基半导体材料被认为是一种潜在的绿色减排技术。为了克服Bi4NbO8Cl半导体中电子和空穴快速复合的缺点,我们通过发展异质结构对Bi4NbO8Cl半导体进行了改性。采用水热法原位合成了含Bi4NbO8Cl的Bi2S3量子点,并用X射线衍射(XRD)、X射线光电子能谱(XPS)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)和紫外-可见漫反射光谱(UV-vis)对其进行了表征。采用Bi2S3/Bi4NbO8Cl异质结构,具有较宽的光吸收区和较低的复合效率,在可见光照射下对有机污染物(罗丹明B,Rh-B)进行光降解。与Bi4NbO8Cl和Bi2S3相比,由于原位合成的Bi2S3量子点能够有效地分离光诱导的电子-空穴对并抑制它们的复合,因此对Rh-B的光催化降解能力更强。此外,DFT数据进一步解释了Bi2S3/Bi4NbO8Cl的界面性质,为新型光催化剂的设计提供了独特的思路,从而促进了其在工业上的潜在应用。 Qu, X., et al. In-situ synthesis of Bi2S3 quantum dots for enhancing photodegradation of organic pollutants. Applied Surface Science, 2020, 501, 144047 实例2:3R-delafossite-CuGaO2合成和光催化性质系列研究 本研究采用简单的低温水热法制备了高结晶度、单相的3R-delafossite-CuGaO2样品。通过实验研究了高锰酸钾的物理化学性质。通过一系列的表征、分析、(光)电化学和光催化活性测试,制备的CuGaO2样品具有产氢和高效光催化降解盐酸四环素(TCH)的潜力。此外,CuGaO2光电极具有长期稳定性和较大的光电流密度。这些观察结果表明,3R-delafossite-CuGaO2是一种潜在的可见光驱动光催化剂。最后,讨论了3R-delafossite-CuGaO2光催化剂可能的突破方向。 Zhao, Q.-M. et al. Delafossite CuGaO2 as promising visible-light-driven photocatalyst: synthesize, properties, and performances. Journal of Physics D: Applied Physics, 2020, 53, 135102 作为高温氧化的一种替代方法,本研究展示了一种基于非平衡水热合成的方法来将过量的O插入3R的delafossite-CuFeO2中。在较低的温度下,或在水热反应的初始阶段,由于水热过程中丙醛的不完全还原,在自组装块层之间残留了大量过量的O。本文在实验观察和理论计算的基础上,不仅证实了过量O的存在并量化了过量O的浓度,而且建立了非化学计量比CuFeO2+δ的生成机理。通常,用常规水热法制备的高结晶度CuFeO2+δ只含有低水平的过量O,但本研究表明,采用微波辅助水热反应可以缓解这一问题。利用密度泛函理论计算了CuFeO2+δ应具有半金属铁磁性和多能隙。通过系统的实验表征和理论计算,阐明了过量氧对材料微观结构的显著影响,同时提高了CuFeO2的电学、光学和电化学性能。由于这些改进,这种材料适用于提高太阳能转化率。光电化学实验和光催化性能的数据证实了这些性质,表明非化学计量比的3R-delafossite CuFeO2+δ化合物在太阳能转化中具有潜在的应用前景。 Liu, Q.-L., et al .Fundamental properties of delafossite […]