
概述
二氧化铈(CeO2)是众所周知的储氧材料,但本文作者研究的新颖之处在于将 CeO2 用于催化CO-SCR脱硝,并引入了过渡金属(TMs)来调节 CeO2 的催化性质。本工作包括:
- 实验合成了 TM–CeO2 样品的结构和这些样品的对 CO–SCR 的催化性能;
- 用 QuantumATK 中的密度泛函理论(DFT)方法阐明了催化剂样品的反应性和选择性;
- 详细报道了催化反应途径,构建了过渡态势能曲线;
- 分析了影响 TM-CeO2(111)表面 CO-SCR 反应的主要因素,讨论了N2O 竞争解吸的选择性。
计算模拟工作
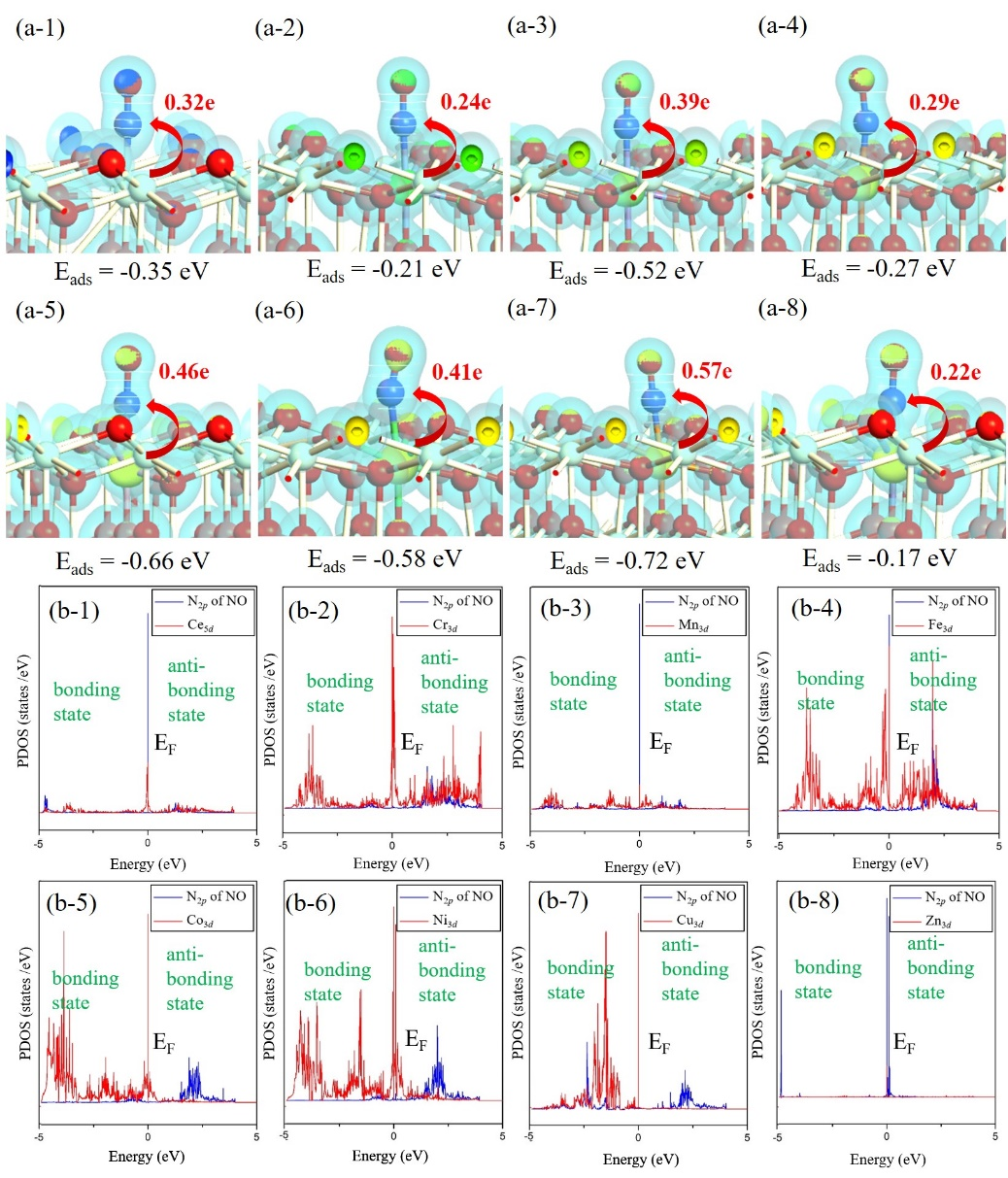
氮氧化物在催化剂表面的吸附。为了研究一氧化氮(NO)分子在 CeO2(111)和 TM–CeO2(111)表面上的吸附能力,作者首先计算了 NO分子在不同表面上的吸附能(图 1a),通过电子密度和电荷分析得到了 NO 在不同表面的电荷转移,并计算了投影态密度验证了吸附态的稳定性。当 Mn、Co、Ni 或 Cu 加入时,TMs 的 d 轨道和 N 2p 轨道之间出现杂化峰,特别是在费米能级以下。
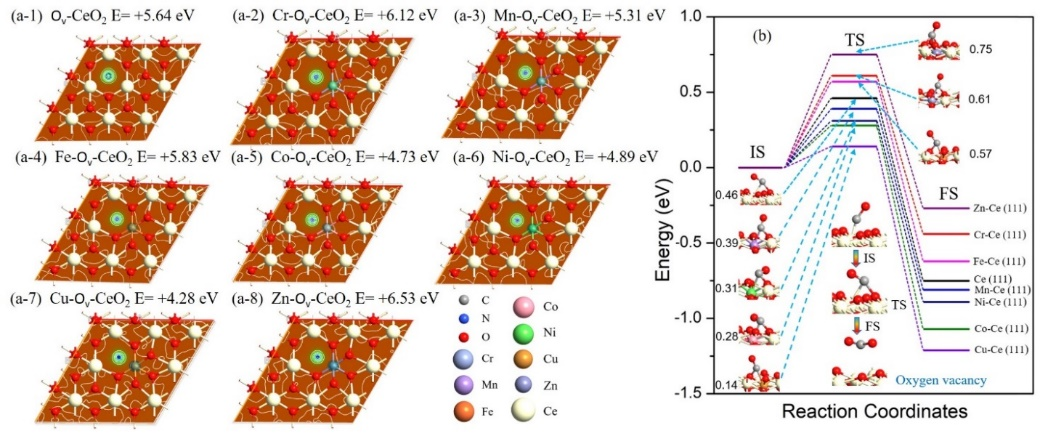
氧空位的形成和作用。氧空位(Ov)在合成或还原 CO 的过程中形成(图2),并且在 *ONN–O 键断裂时充当氧受体。作者讨论了 TM–O–Ce 中心的 Ov 形成能。金属氧化物催化剂上的氧化还原反应通常由 Mars–van Krevelen 机制解释。在氧空位形成后,电子首先在 Ov 处聚集,然后迁移到周围的金属位。由于晶格氧更容易接受电子,电子在晶格氧的作用下发生迁移和扩散。同时,Cu-CeO2(111)更容易产生氧空位,产生更多的电子。
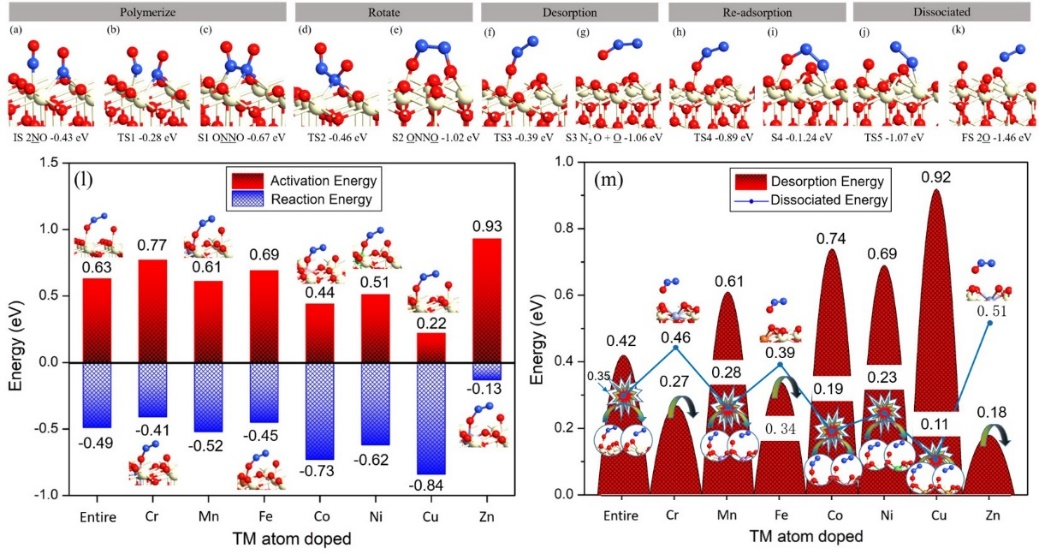
催化反应路径与能量。图 3a–f 展示了通过 *ONNO 中间产物的离解生成 N2O 和 *O 物种。*ONNO 的能量收益 ΔE 为−0.24 eV。π* 反键作用激活 NO 的 Ea 为 0.15 eV,足以引发二聚反应。当 N–O 键伸展时,ONNO 容易分解为 N2O 和*O,其中 Ea=0.63 eV。两个 NO 分子被 ONNO 中间体还原为 N2O,从而显示 N2O 在 TM-CeO2(111)表面还原为 N2。图 3g–k 显示了 N2O 在 CeO2(111)表面上的反应途径。N2O 的 O 端位先吸附在 CeO2(111)表面,N 端位再吸附,最后与能量较弱的 *NNO 发生翻转。物种分解成一个 N2 分子和 O 原子。
参考
- Zhisong Liu, Feng Yu* Dong Dong, Rongrong Gui, Wenjian Li, Ruobing Sun, Yinji Wan, Jianming Dan, Qiang Wang*, Bin Dai*. Transition-metal‐doped ceria carried on two-dimensional vermiculite for selective catalytic reduction of NO with CO: Experiments and density functional theory. Appl. Surf. Sci.(2021) 566, 150704, DOI: 10.1016/j.apsusc.2021.150704.