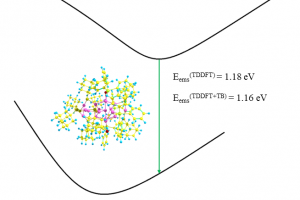
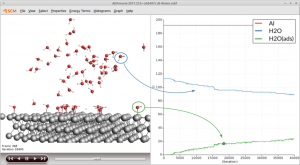
近年在分子动力学模拟,尤其是涉及化学反应的分子动力学模拟、燃烧与裂解领域,AMS 成为成功应用的佼佼者,其高速计算、实用的分析功能,得到用户广泛认可。在力场参数拟合、机器学习方面也被研究者们迅速运用起来。AMS 一直保持着强劲的创新,在传统量子化学、第一性原理材料计算、OLED 器件模拟等领域持续改进。 OLED Workflow 生成不同材料的参数,作为 Bumblebee 的输入,以模拟 OLED 像素特征与性能 计算提速 5 倍 图形界面调用 Lammps ADF – 量子化学 ROKS-TDA 限制性开壳层的激发态计算 精确 IP 值计算:顶点校正 GW 方法 基于 ROSE 的激子转移积分 基于片段轨道的 CDFT,能够有效将电荷局限到体系的某个局部,并支持此类体系的分子轨道计算、能级计算、EDA 分析、ETS-NOCV 分析、转移积分计算等 自旋极化>0的体系,SOC的计算能够自动收敛到正确的自旋态上 机器学习 最新的机器学习势:CHGNet, FairChem, MACE, MatterSim, ORB, 以及更多 基于其他 Python 环境的 ASE 计算器 图形界面 新的 Packmol 界面:非均相体系支持设定非固体区域的密度、支持按摩尔比填充, Jupyter Notebooks 图形界面响应速度大幅度提升,大体系显示流畅度大幅度提升 Quantum ESPRESSO […]