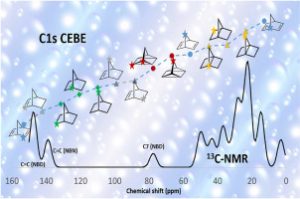
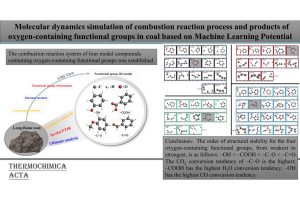
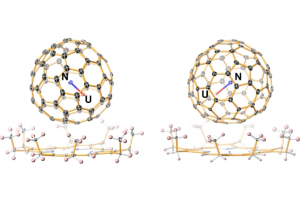
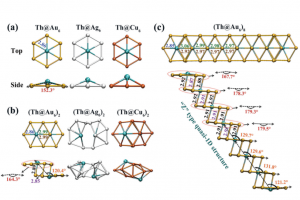
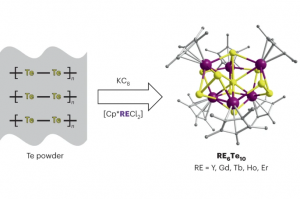
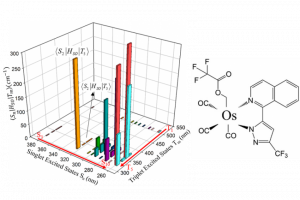
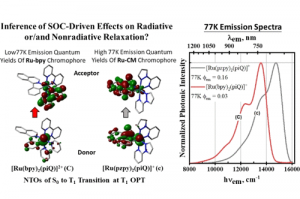
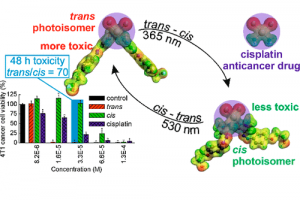
ReaxFF分子动力学研究α-CPP和β-CPP燃料的热解机理(天津大学) 具有高密度和高比冲的新型燃料分子是航空航天燃料的理想替代品,了解它们的燃烧或热解化学是成功应用的必要条件。作者使用 ReaxFF MD 研究了新型燃料分子环丙烷化 α-蒎烷(α-CPP)和环丙烷化 β-蒎烯(β-CPP)的热解。结果表明,燃料分子的初始热解主要通过三个步骤完成:三元环中的 C 单键断裂形成二自由基,四元环发生开环反应形成内部具有环状结构和双键的二自由基;六元环向三烯烃分子或具有不饱和键的二自由基开环。在 α-CPP 和 β-CPP 的初始反应中, C11H18 → •CH3 + •C10H15 和 C11H18 → •C6H9 + •C5H9 分别占主导地位。进一步分析表明,C5H9 和 C6H9 自由基是热解过程中的重要中间体。动力学计算表明,α-CPP 和 β-CPP 分子的活化能分别为 51.44 kcal/mol 和 49.14 kcal/mol,小于 JP-10、RP-1和其他燃料的活化能。这项工作为在原子水平上揭示高能燃料分子 α-CPP 和 β-CPP 热解的整体反应机理提供了理论基础。 Pyrolysis mechanism of α-CPP and β-CPP fuels by ReaxFF molecular dynamics, Menghui Chen, etc., Computational and Theoretical Chemistry, 2024, Volume 1240, […]