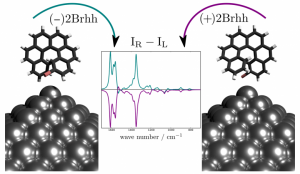
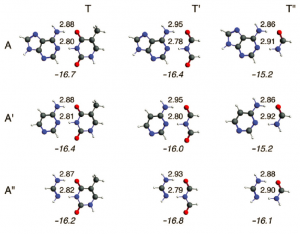
参考文献: D.V. Chulhai and L. Jensen, Simulating Surface-Enhanced Raman Optical Activity Using Atomistic Electrodynamics-Quantum Mechanical Models, J. Phys. Chem. A, 118, 9069 (2014) 拉曼光学活性已经成为探测有机小分子和生物分子几何结构的有力工具。但是作为一项新技术表面增强拉曼光学活性(SEROA)仍然是实验方面的一个挑战。小分子的SEROA已经能够通过ADF,将速度度规响应张量应用到离散相互作用模型/量子力学(DIM/QM模型)来模拟。在这种模型中,纳米粒子被当做相互作用的原子极化子的集合体来处理。计算结果能够揭示局域电场(local electric field)、电场梯度强度、等离激元宽度,以及分子朝向如何影响SEROA信号。