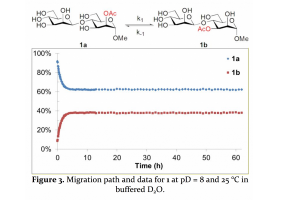
文献资料:Robert Lassfolk, Jani Rahkila, Mikael P. Johansson, Filip S. Ekholm, Johan Wärnå, and Reko Leino, Acetyl group migration across the saccharide units in oligomannoside model compound, J. Am. Chem. Soc. 2019, 141, 4, 1646-1654 乙酰化低聚糖在自然界中很常见。虽然它们参与了一些生化和生物过程,但乙酰基的作用及其迁移的复杂性并没有引起人们的注意。通过有机合成、核磁共振波谱和量子化学模拟相结合的方法,作者发现乙酰基迁移比以往所知更为复杂。利用合成的寡糖苷模型化合物,首次证明了乙酰基在低聚糖和多糖中的迁移可能并不限于在一个单糖单元内部,还可能涉及到两个不同的糖单元之间的糖苷键上的迁移。观察到的这一现象不仅是非常有趣的化学问题,而且也对酰化碳水化合物在自然界中的潜在生物学作用提出了新的问题。 本文在CCSD(T)/COSMO-RS级别进行了流体热力学计算。实验观察到1a与1b的比率约为60:40,COSMO-RS模拟得到的比率为63:37(ΔG = +1.3 kJ/mol)。其中流体热力学计算由AMS中的COSMO-RS模块完成。