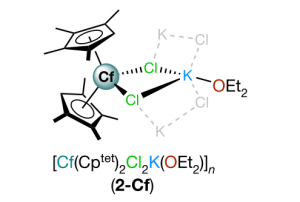
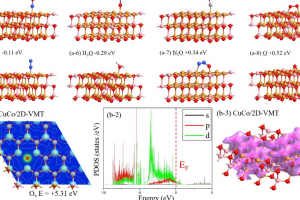
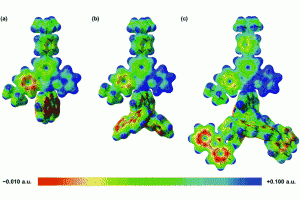
四川大学化学学院苏静教授课题组、美国洛斯阿拉莫斯国家实验室锕系化学实验和理论研究团队,对锕系元素锎(Cf)的研究取得重要进展。锎(Cf)的价电子参与化学成键的能力、自旋-轨道耦合在电子结构中的作用以及反应类型等化学性质,缺少相关研究报道。该研究首次合成并表征了一种二茂型锎配合物[Cf(C5Me4H)2Cl2K(OEt2)]n (如下图所示),特别是对Cf元素的化学成键和光谱性质进行了深入的理论研究。 为研究其化学成键特点,通过AMS软件ADF模块,对[Cf(C5Me4H)2Cl2K(OEt2)]n及其镧系类似物[Dy(C5Me4H)2Cl2K(OEt2)]n进行了分子轨道能级、分子轨道组分、电子Mulliken布居、键级以及QTAIM等分析,发现Cf-C键以离子键特征为主,共价成分非常微弱。 结合ADF的含时密度泛函理论(TDDFT)计算分析,解释了深橙色化合物[Cf(C5Me4H)2Cl2K(OEt2)]n与无色化合物[Dy(C5Me4H)2Cl2K(OEt2)]n在颜色方面的差异来源:[Cf(C5Me4H)2Cl2K(OEt2)]n中配体价轨道(激发对应的占据轨道)与Cf 5f空轨道(激发对应的空轨道)的能隙更小,配体到金属的电荷转移跃迁主要发生在可见光范围;Dy 4f空轨道能级较高,从而[Dy(C5Me4H)2Cl2K(OEt2)]配体价轨道与Dy 4f空轨道能隙更大,相关激发能对应到了紫外区域,从而呈现出不同颜色。 作者还采用高精度多参考态从头算方法CASSCF/NEVPT2,并考虑旋轨耦合作用,精确计算了锎化合物的UV-vis-NIR光谱,得到了与实验一致的结果。 在重元素,尤其是超重元素、锕系化学领域,理论研究中涉及的相对论效应、基函数精度尤为重要,AMS-ADF包括目前最先进的全电子相对论方法以及高精度STO基组,是超重元素研究领域中尤其得力的理论研究工具。其EDA、ETS-NOCV方法在研究金属配合物成键方面,能提供非常清晰的、定性与定量的化学成键图景。 参考文献 Goodwin, C.A.P., Su, J., Stevens, L.M. et al. Isolation and characterization of a californium metallocene. Nature 599, 421–424 (2021). https://doi.org/10.1038/s41586-021-04027-8