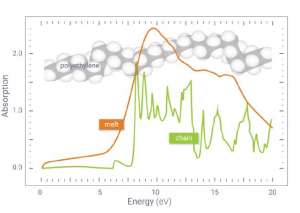
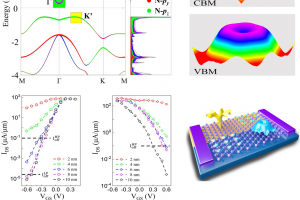
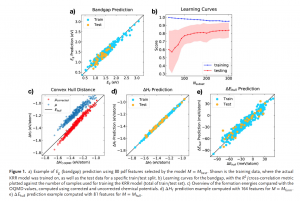
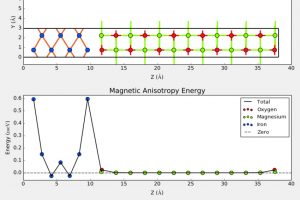
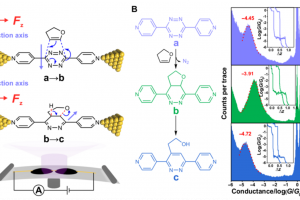
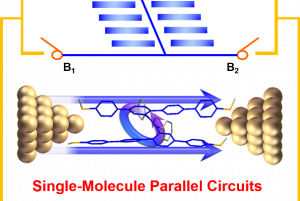
概述 光伏材料与器件是实现太阳光能直接转化为电能的一种重要途径,更广泛的光电转换材料与器件则可以应用于光信号传感器等领域。基于半导体材料构造的异质结构是这类材料应用的关键,一般来说影响光电转换效率的因素有:材料吸光能力、电子空穴对分离效率、载流子迁移效率。使用 QuantumATK 可以方便的研究: 多层结构建模高级的多层结构界面建模工具,支持直接构建任意层数的多层结构,控制每层的取向等复杂体系的结构优化和动力学对超大体系使用机器学习力场(MTP)进行优化机器学习模型支持对缺陷、界面和无定形结构进行训练半导体材料的准确带隙和光吸收 包含超快的HSE06杂化泛函方法和快速的k空间采样方法,可以轻松计算千原子体系 半导体材料的载流子有效质量和迁移率 采用完整的电声耦合方法 半导体材料的点缺陷直观、智能的点缺陷建模工具计算点缺陷的形成能和缺陷捕获能级自动化的缺陷扩散动力学研究工具半导体异质结构的能带排列 采用独特的界面模型直接计算,不必考虑真空能级校正等复杂处理 异质结器件的光生电流和开路电压 直接得到光电流谱和太阳光照下的电流密度可以直接考虑光电转换过程中的温度效应 应用案例 实例1:探索钙钛矿锡基光伏太阳能电池性能的非平衡格林函数与宏观途径 此项研究使用两种计算方法来研究混合钙钛矿型锡基光伏太阳能电池。第一种方法基于电子输运性质计算,结合了密度泛函理论和非平衡格林函数理论。作者研究了透射谱和态密度。结果表明,由于电子态的离域化,从 MASnI3 到 MASnBr3 的输运带隙减小,表现出较大的电子输运能力。第二种方法是基于漂移扩散方法模拟,发现钙钛矿-锡基混合光伏电池的参数明显依赖于钙钛矿吸收层的厚度。表面复合速度在 1~10 cm/s 和 102~103 cm/s 范围内,MASnIBr2 和 MASnBr3 的效率分别达到 16.07% 和 12.52%,可以提高太阳能电池的性能。 详见: 探索钙钛矿锡基光伏太阳能电池性能的非平衡格林函数与宏观途径Physica B: Condensed Matter, Elsevier BV, 2020, 591, 412247 实例2:二维材料的结构电子态模拟 论文作者对实验上得到的多层结构进行了模拟,得到了与实验一致的结果。文中使用DFT-D2范德华力泛函(注:QuantumATK最新版现已支持DFT-D3)对结构进行了优化,用GGA进行了结合能的计算,使用MetaGGA进行了精确带隙的计算。作为比较,又使用HSE06杂化泛函的DOS计算。注:新版的QuantumATK中LCAO基组已经支持HSE06杂化泛函,并能够进行超快的电子态计算。 详见: 用石墨烯包裹实现二维氮化镓(Nature Materials, 2016)超快速的HSE杂化泛函计算半导体准确带隙 实例3:机器学习研究钙钛矿光伏材料的稳定性和带隙 钙钛矿的成分调控让人们能够精确控制其在光伏应用所需的材料性能。然而,同时解决效率、稳定性和毒性仍然是很大的挑战。混合的无铅钙钛矿和无机钙钛矿最近显示出了解决此类问题的潜力,不过它们的组分空间巨大,即使采用高通量方法也很难发现有希望的候选结构。此项研究通过使用由密度泛函理论生成的344个钙钛矿的新数据库,使用与元素无关的通用指纹信息的机器学习方法可以快速而准确地预测关键属性。使用验证子集预测的带隙、形成能、和凸包距离分别在146 meV、15 meV/atom 和 11 meV/atom 内。得到的模型可以用于预测完全不同的化学组分空间中的趋势,并进行快速的组成和结构空间采样,而无需进行昂贵的从头算模拟。 详见: […]