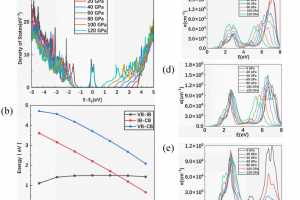
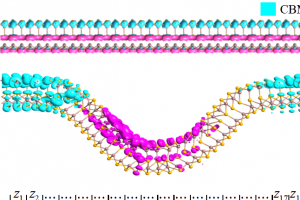
研究背景 随着全球能源需求的持续增长与环境问题的日益严峻,开发高效率光电转换材料成为新能源领域的重要研究方向。传统单结半导体光伏器件受 Shockley–Queisser 极限的限制,其光电转换效率难以进一步提升。为突破这一理论瓶颈,中间带(Intermediate-Band, IB)光电材料被提出,通过在禁带中引入中间能级,使材料能够吸收低能光子,从而显著提高光谱利用率和器件效率。然而,实现理想中间带结构仍面临巨大挑战,尤其是在保证中间带与价带、导带有效分离及适当占据的条件下。 金刚石作为一种典型的宽禁带半导体,具有优异的热导率、化学稳定性和载流子迁移率,是构建高性能光电器件的潜在候选材料。然而,其超宽带隙限制了对可见光的吸收能力。近年来,缺陷工程被广泛用于在半导体中引入中间能级,但单一缺陷调控往往难以实现理想的中间带结构。同时,应变调控(应力工程)作为一种有效的能带调节手段,可进一步调控电子结构与能级分布。 因此,将缺陷工程与应变调控相结合,系统研究其对金刚石中间带形成及光电性能的影响,对于实现高效中间带光电材料具有重要意义,并为新型高效率光伏器件的设计提供理论依据。 研究内容 该研究围绕在金刚石中构建理想中间带光电材料这一目标,系统探讨了缺陷工程与应变调控协同作用对其电子结构的影响。首先,基于第一性原理计算,构建多种含缺陷的金刚石模型,分析其对能带结构和态密度的调控作用,重点考察缺陷在禁带中引入中间能级的能力及其位置分布。 图 1.(a)3 × 3 × 3 超胞中 B-As 共掺杂金刚石的构型(b)3 × 3 × 3 超胞中 B-As 共掺杂金刚石的能带结构(c)2 × 2 × 2 超胞中 B-As 共掺杂金刚石的构型(d)2 × 2 × 2 超胞中 B-As 共掺杂金刚石的能带结构(粉色球代表 B 原子,紫色球代表 As 原子) 在此基础上,进一步引入应变调控,研究外场对中间能级位置、带宽及占据情况的影响。通过系统比较不同缺陷类型与应变条件下的电子结构特征,评估是否满足理想中间带材料的关键要求,即中间带应位于禁带中间、与价带和导带保持有效分离,并具备合适的电子占据状态。 图 2.(a)未受压 C61B2As 的结构示意图和态密度图(b-d)分别为沿 X、Y 和 Z 方向施加 80GPa 单轴应力时 C61B2As 的结构示意图和电子态密度图(箭头表示压缩方向) 进一步,通过计算不同单轴压应力下 C61B2As 的态密度、带隙以及光吸收系数,研究单轴应力对 C61B2As 电子、光学性质的影响。 图 3.(a)沿 Z […]