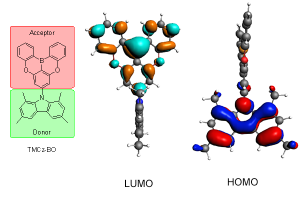
传统上,OLED器件只有25%的内部量子效率,因为只有单个激发态可以用来发射荧光,另外75%激发到三重态。近年来重金属的强自旋轨道耦合(SOC)效应被用来增强系间窜跃,将理论量子效率提高到100%。使用重金属的一个缺点是,但是金属配位键较弱,导致蓝色OLED的寿命较低。另一个方案是使用热激活延迟荧光(TADF)机制,通过逆向系间窜跃(RISC)将最低三重态(T1)转换为最低单重态(S1)。虽然TADF机制很有前途,但如果逆向系间窜跃发生得不够快,它可能会受到其它竞争进程的阻碍。这需要较小的S1-T1能隙,以及S1态和T1态之间良好的振动耦合,自旋轨道耦合(SOC)对系间窜跃速率也有很大的影响。 日本九州大学OPERA究中心发现了一种新的深蓝色TADF发射器TMCz-BO,在467nm处高效发光。文章使用AMS-ADF考虑自旋轨道耦合的TDDFT(SOC-TDDFT)的计算,解释了TMCz-BO的TADF发光为何如此高效:T2和S1状态之间的旋轨耦合矩阵元(SOCME)相对较大(0.124cm-1),有利于逆向系间窜跃,而系间窜跃回到T1状态,则由其0.001cm-1的低SOCME所阻碍。 这种计算策略对TADF发射体的进一步发展和分子设计具有重要意义。用AMS-ADF很容易优化基态和激发态。所用到软件功能的中文教程参考:第三代OLED材料性能的第一性原理计算:通过DFT预测TADF逆向系间窜跃速率 (JACS, 2017) Jong Uk Kim, In Seob Park, Chin-Yiu Chan, Masaki Tanaka, Youichi Tsuchiya, Hajime Nakanotani & Chihaya Adachi, Nanosecond-time-scale delayed fluorescence molecule for deep-blue OLEDs with small efficiency rolloff, Nat Commun 11, 1765 (2020)