
最近Kaupp课题组结合实验和理论,对过渡金属磷化物化学进行了详细研究。用核磁共振监测Pt-PPh3与Pt(0)配合物的反应,并用DFT计算确证了反应中间体的结构,该研究有助于理解C-P键形成和断裂的催化过程。
研究表明,Pt(0)能够可逆地插入到三苯基膦的邻位C-H中,形成双核μ-氢化物配合物。对于这种亚稳态配合物,ADF计算有助于明确最有可能的同分异构体,以匹配实验中测量的化学位移和耦合数据。由于该配合物与反应物处于平衡状态,最终通过一个磷-苯基键的C-P键断裂形成热力学上有利的磷酰亚胺产物。
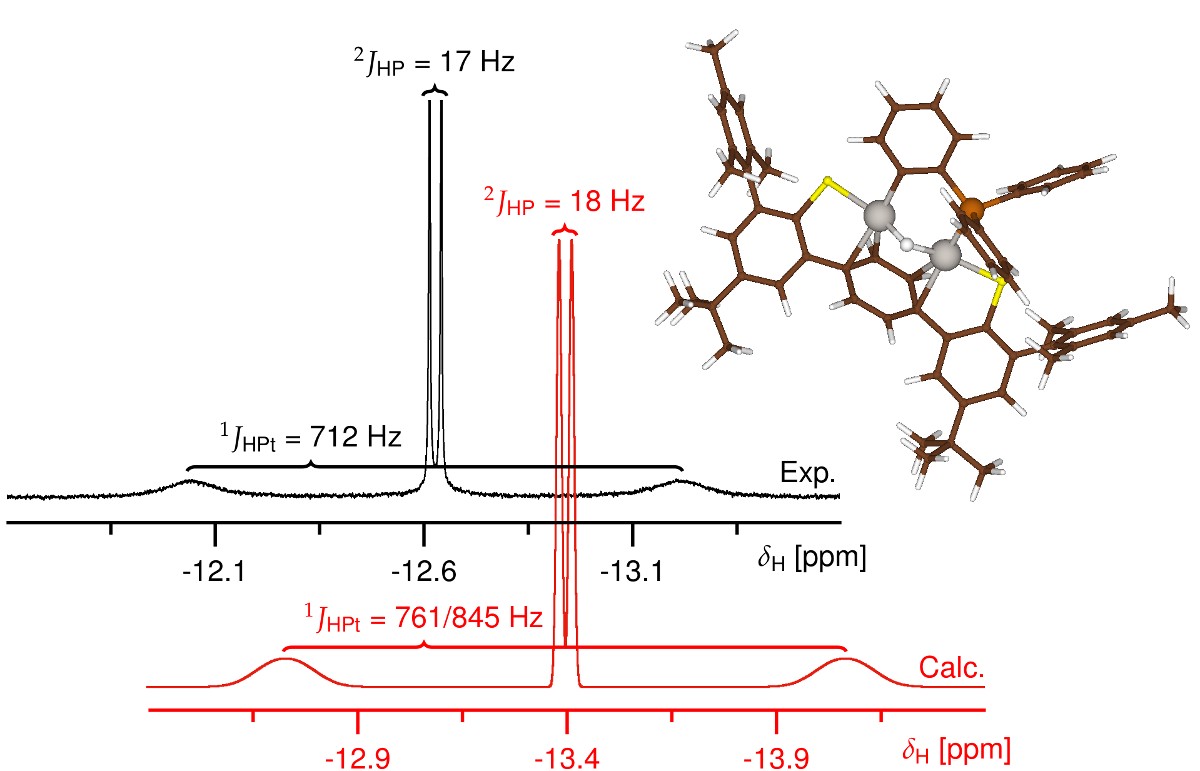
AMS中ADF模块结合ZORA相对论方法以及COSMO溶剂化模型,计算得到一致性非常好的1H、31P,以及195Pt NMR位移与耦合
进一步研究C-P插入产物的氧化还原化学,EPR测量结果表明发生氧化还原反应的是硫醇基而不是金属中心。ADF的g因子与A张量计算结果,完全支持这些实验数据。
Berkefeld, A., Reimann, M., Hörner, G., Kaupp, M., Schubert, H.. C–P vs C–H Bond Cleavage of Triphenylphosphine at Platinum(0): Mechanism of Formation, Reactivity, Redox Chemistry, and NMR Chemical Shift Calculations of a μ-Phosphanido Diplatinum(II) Platform. Organometallics 39, 443-452 (2020).