Index of Topics Our Comprehensive Entertainment Library Superior Safety & Transparent Play Protocols Payment Methods & Transfer Time Extensive Loyalty Program Mobile Gaming Platform Assistance Channels & Help Welcome to the platform, in which advanced technology merges with outstanding gambling experiences. At PJSpins high roller, we’ve built a reputation on delivering excellent fun worth by […]

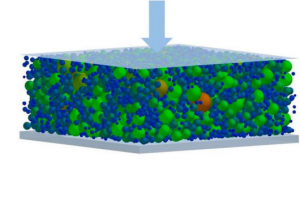
离散元方法和通过 X 射线计算机断层扫描表征的锂离子正极结构电化学建模
概述 锂离子电池因其高容量、高功率的优点,在储能领域发挥着至关重要的作用,广泛用于电动汽车和便携式电子产品等。电动汽车的快速发展要求高能量密度、高循环寿命和低成本,进一步改进制造工艺和电极设计仍然存在挑战。 在电极制造过程中,压延工艺是定制电极微观结构的关键步骤。在本研究中,使用离散元法(DEM)和粘结颗粒模型研究压延过程对电极微观结构的影响,对使用 X 射线计算机断层扫描(XCT)表征的真实电极结构和理想化 DEM 结构进行综合评估,计算分析基于断层扫描和 DEM 的电极结构及输运特性,即孔隙率分布、比表面积和曲折因子。在考虑碳粘合剂域(CBD)相之后,进一步分析电化学性质。 亮点 对基于高分辨率 XCT 表征和 DEM 的锂离子电池阴极结构进行评估在 Simpleware 软件中生成高质量的四面体网格,将网格模型导入 COMSOL 中进行仿真考虑不同的压延水平和 CBD 相,分析电化学性能 方法 图像处理 电极结构由 96 wt% LiNi0.6Mn0.2Co0.2O2(NMC622,BASF)、2 wt% C65 炭黑(Imerys)和 2 wt% PVDF(Solvay)配制,使用带有 MTI MSK-HRPMR100DC 压延设备的辊压机将干燥的电极压延两次。通过 XCT 对阴极结构进行表征,采用未压延结构和压延结构与 DEM 预测进行比较。使用基于机器学习的图像分析工具 Ilastik 预测扫描图像中不同相的体积分数,即 AM 颗粒相、CBD 相和宏孔相。 原始扫描图像(图a)经过过滤和二值化后获得 AM 颗粒相(图b),进行分割处理分离和标记单个 AM 颗粒(图c),并获得其各自的体积和坐标。在 DEM 模拟中,阴极结构内的颗粒近似为球形颗粒(图d)。 图1:AM 颗粒相的图像处理步骤 DEM […]
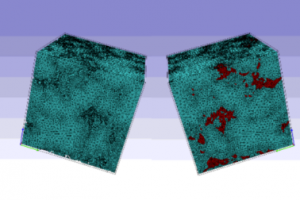
利用数字岩石计算有效物理性质的形态学变换策略
概述 岩石是一种天然多孔介质,其结构中不仅包含岩石骨架,还有大量不规则的孔隙和孔隙流体。储层岩石的电学、力学和物理性质评价对于油气勘探具有重要意义。 基于 micro-CT 图像的数字岩心技术为岩石物理研究提供了新的途径,与传统实验相比也具有很多优势。利用三维数字岩心模型和多物理场模拟可以对岩石样品的有效物理性质进行数值评价。然而,在利用扫描图像进行数字岩心建模的过程中,存在各种影响岩石性质数值计算精度的因素,如形态学变换等。 应用于同一岩心样品的形态学变换策略会导致不同的孔隙率计算结果。研究表明,图像处理算法也会影响孔隙结构的重建,进而对样品的电学性质计算产生较大影响。本项目采用不同的形态学变换策略,对体素为 6003 的三维数字岩心模型进行研究。通过结合使用 Simpleware 和 COMSOL Multiphysics 软件,计算测量岩心模型的孔隙率和孔隙结构,模拟岩心模型的有效电学性质。 亮点 研究对象包含砂岩、页岩和碳酸盐岩在 Simpleware 中使用不同的形态变换方案为每个岩石生成多个微观结构在 COMSOL 中计算岩心模型的有效弹性模量和介电常数 数字岩心建模 获取图像数据 使用蔡司 Xradia 520CT 扫描设备对页岩、砂岩和碳酸盐岩样品进行扫描,它们的尺寸(8 mm)和孔隙度范围(4%-25%)均相同。页岩样品具有明显的层状结构,砂岩样品具有多种孔隙类型、孔径、形状和分布,而碳酸盐岩样品孔隙率小且孔隙结构简单。 图1:micro-CT 扫描样品的横截面:砂岩(左)、碳酸盐岩(中)和页岩(右) 图像处理和分割 扫描获得的灰度图像中存在着噪音,需要通过滤波器提高信噪比。将扫描图像导入 Simpleware 后,首先应用中值滤波器(Median filter)改善图像。为更好地区分孔隙和骨架,图像分割的阈值选择也非常重要。鉴于实测孔隙率已知,可以采用公式基于岩心孔隙率的最佳分割阈值进行分割。 基于图像的三维重建 理论上数字岩心尺寸越大,对岩石微观孔隙结构和宏观特征的表征就越准确。但这样对计算机资源和性能的要求很高。经多次测试发现,当数字岩心尺寸为600 × 600 ×6 00 体素时,物理性能(如孔隙率、弹性模量等)受到的影响最小。因此,本研究选择此尺寸作为代表单元的体积。 在 Simpleware ScanIP 中对图像进行重建、处理和分割后,应用不同的形态学变换策略:ED 腐蚀和膨胀,OC 打开和闭合,SC 平滑滤波器和孔洞填充,FI 填充间隙和孤岛移除。在 Simpleware FE 中对处理过的模型进行网格划分,生成高质量的网格模型并导入 COMSOL 中进行仿真。 图2:形态学变换的结果 图3:数字岩心网格模型:孔隙(左)、骨架(中)、孔隙和骨架(右) 形态学变换对岩石性质的影响 孔隙率 不同形态学变换对岩石孔隙结构的敏感度不同。ED 和 OC 对连续性孔隙的影响更大,而 […]
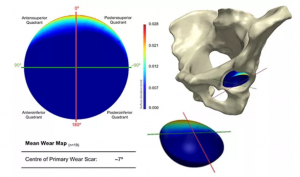
构建回收髋关节植入物中髋臼磨损在体内位置的统计形状模型
概述 已知髋臼杯的边缘磨损与更大的材料体积损失有关,但这种磨损模式在体内的位置却尚不清楚。本研究在 Simpleware 软件中开发了一个使用 CT 成像、检索分析和统计形状模型(SSM)识别体内最常见磨损位置的工作流程。 回收 20 个在平均 90 个月后翻修过的金属对金属髋臼表面,进行形状方差分析。还研究了磨损量、植入位置、时间、尺寸和性别对体内磨损位置的影响,有助于更好地理解髋关节植入,为将来的产品设计和植入物定位中安全区域的细化提供重要信息。 亮点 对 20 个回收的金属对金属髋臼表面进行扫描,观察磨损模式。使用 Simpleware 软件识别体内生成最常见的髋臼磨损模式特征,构建统计形状模型(SSM)。SSM 可以提升对髋关节植入物功能的理解,从而为未来的设计决策提供信息和细化植入物定位的安全区。 介绍 髋关节置换术中轴承表面的机械磨损会影响临床表现,导致功能受损和有害碎片的释放。由于金属对金属(MOM)髋关节置换失败的发生率很高,需要开展关于这些表面上的磨损程度和确定髋臼杯边缘磨损与大量磨损碎屑间关系的研究。虽然 MOM 植入物目前很少应用于髋关节植入,但仍能为分析髋关节置换术的力学提供有价值的数据。此外,尽管已知髋臼边缘磨损发生在体内,但对其在髋臼腔内的位置了解较少。 统计形状模型(SSM)为描述相关几何结构的形状和位置提供了一种有价值的方法,特别是在分析解剖特征时可以将平均形状和形状方差可视化。本研究的目标是通过结合 CT 成像和检索分析技术在 Simpleware 软件中创建一个 SSM,用于识别髋关节置换术中髋臼组件最常见的体内磨损模式。 图1:研究设计和工作流程及 Birmingham 髋臼组件背侧表面和其不对称的鳍状物(红色部分) 根据纳入标准选择 20 例 MOM Birmingham 髋关节置换(BHRs)的髋臼组件进行研究,需要在取出前获得未翻修骨盆和植入物的 3D CT图像。研究人员对植入物进行修复,在体内的平均时间为 90 个月,手术原因是对金属碎片的不良反应、不明原因的疼痛或无菌性松动。 使用定制的软件解决方案(Robin 3D)计算每个 BHR 的体内位置,采用骨盆前平面(APP)作为标准化坐标系,并报告解剖倾斜度和前倾角的值。然后使用卡尔·蔡司的坐标测量机(CMM)将每个回收臼杯铰接表面的几何形状捕获为点云,同时通过经验证的自动化软件解决方案计算每个髋臼表面的材料损失体积。 Simpleware 中的分割和配准 将翻修前的体内植入物 CT 图像以 DICOM 文件格式导入 Simpleware 软件,使用半自动化工具分割生成植入物和骨盆模型,通过处理减少金属伪影的同时保留几何精度。然后将球体与股骨头贴合,利用布尔运算从植入物模型中分离出髋臼杯,平面与臼杯边缘完全贴合用于去除模型中相对较差的部分,包括股骨钉的剩余物。由 CMM 数据生成髋臼杯的开放表面并以 STL 文件导入 Simpleware 软件,使用半自动化工具将其与孤立的臼杯模型配准,对齐稳固的鳍状物。随后对配准的髋臼杯表面和骨骼模型进行镜像和适当缩放。 图2:(1)将髋臼表面(紫色)与从 3D […]

开源插件程序:在三维建模中赋予皮质骨和骨小梁不同的材料属性
概述 骨小梁是一种海绵状的各向异性材料,将载荷从关节表面转移到更为致密的皮质骨。皮质骨的密度和刚度更为一致,处理重复的拉伸和压缩负载产生的压力。在模型中准确地定义这些材料的特性对于将医疗器械设计和手术原理转化为临床应用至关重要。骨科医疗器械通过提供骨骼愈合或关节功能的环境帮助患者康复,外科医生需要根据患者的骨质量、合并症及自身经验,为最佳植入物做出最准确的预测。 精准的计算模型可以使外科医生和医疗器械工程师能够模拟患者骨骼内每种植入物类型的性能,以便就植入物的设计、选择和手术技术做出更为明智的决策。本研究开发了一个利用骨矿物质密度通过定量计算机断层扫描(QCT)确定有限元分析(FEA)中材料属性的插件程序,与图像处理软件 Simpleware 结合简化了该方法的工作流程,可更方便于临床和研究的应用。 亮点 使用 Simpleware ScanIP 的图像处理和 Python 脚本编辑工具对 CT 扫描数据进行预处理。将单个 CT 体素的灰度值转换为杨氏模量开发的插件程序(PIP)以开源的方式共享 灰度值转换为杨氏模量的流程 输入骨小梁/皮质骨的截断密度及相应的灰度和 QCT 密度值将医学数字成像和通信(DICOM)格式的 QCT 值转换为湿表观密度湿表观密度值根据相应的组织转换为杨氏模量使用调整后的 DICOM 文件创建 3D 模型 方法 将扫描的体模 DICOM 图像数据导入 Simpleware 软件中,经过图像处理后分割为包含每个样本的圆柱体/圆盘。使用 Simpleware ScanIP 的灰度测量功能获取每个样本的平均灰度值,以 HU 为单位测量并将结果转换为 DICOM 存储的灰度值。通过对整个体模体积的 HU 进行采样(需要定义为组织等效电子密度样本),可以减少噪声的影响,并且结果可以更准确地表示整个体模的测量放射密度。 图1:研究体模中包含伪影的 DICOM 图像 使用 ScanIP 的 Profile Line 测量工具对每个灰度值进行采样,通过在样本中画线即可导出其灰度值。启动插件程序(PIP)后,提示输入灰度值和体模对应制造商定义的 QCT 密度,跟每个用户的 CT 扫描仪和体模相关。 PIP […]
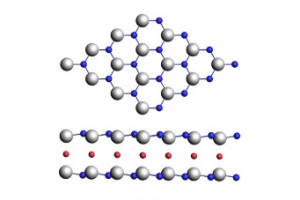
非金属原子嵌入二维 GaN 双层中的半金属性研究(Appl. Phys. Lett. 2023)
摘要 半金属材料是高效自旋电子器件的理想工质。常见的半金属材料大多含有过渡族金属,局域的d或f电子使其可能不适合未来的自旋电子学应用(如基于碳和有机分子的生物相容性电子器件和设备)。为了克服这一问题,人们提出了无过渡族金属的p轨道半金属材料,但是这些材料的制备往往需要严苛的条件,例如强外场或高载流子掺杂。南京大学陆海鸣副教授课题组与伊犁师范大学黄以能教授合作,采用第一性原理计算发现F插层的GaN双层(F-Ga2N2)是一种不受插层原子浓度影响的p轨道半金属材料,且其半金属性在双轴应变-10%~10%时都可稳定存在。该研究成果为未来自旋电子学应用提供了一种设计无过渡金属的半金属材料的新策略。 图1、F-Ga2N2的(a)几何构型、(b)PBE计算得到的能带图和(c)总态密度和分波态密度 通过第一性原理计算,作者发现F原子倾向于插层在上下两层Ga原子的中间(图1a),从能带图(图1b)可以看出自旋向上部分具有较大的带隙而自旋向下部分则是越过了费米面,因此F-Ga2N2呈现出了半金属性质,且半金属带隙高达0.688 eV。此处能带、态密度的计算,参考中文教程:https://www.fermitech.com.cn/wiki/doku.php?id=adf:halfmetallicity 结合分波态密度图(图1c)可以看出该材料的半金属性主要来自于N原子的p轨道,这可以解释为:F原子的插层弱化了Ga原子和N原子间的键合,增加了N原子的p轨道能量,从而使得本征GaN双层的价带顶上移越过了费米面。 图2、无磁态F-Ga2N2的(a)能带图和总态密度和分波态密度图 为了理解这种不同寻常的p轨道半金属性,我们首先计算了无磁态F-Ga2N2的能带图和态密度图(图2),得出其Stoner准则为3.445,表明该材料具有巡游铁磁性。构建4*4*1超胞后的铁磁态、反铁磁态和无磁态能量计算也证实了其铁磁性基态。 图3、不同插层浓度的F-Ga2N2体系的能带图 既然吸附/掺杂浓度通常会影响材料的电子结构,我们也研究了插层浓度对F-Ga2N2半金属性质的影响。当体系的插层浓度从前述100%降低到25%、11%和6.25%时,从图3可以看出插层体系的半金属性一直存在。 在器件设计中,二维材料通常由衬底支撑,两者之间的晶格失配引起的应变也会对二维材料的磁基态和电子结构产生影响。通过计算我们发现,F-Ga2N2的磁基态在双轴应变-10%~10%之间都没有发生变化,而且在10%的双轴拉伸时该体系的半金属性依然存在。 图4、不同双轴应变时的反铁磁-铁磁能量差DE = EAFM–EFM以及拉伸10%的能带图体系的能带图 总结 本文利用AMS软件的BAND模块,进行了F原子插层改变GaN双层电子结构和磁基态的研究。研究发现,不含过渡族金属的F-Ga2N2体系表现出了p轨道半金属性,其中部分填充的平带诱导的Stoner不稳定导致了铁磁有序,而强自旋劈裂导致了半金属性的出现。0.688 eV的半金属带隙使得该材料的半金属性在高温下依然可以存在。此外,鲁棒的半金属性不受插层原子浓度影响,且其在-10%~10%的双轴应变下也不会被破坏。 参考文献: Bai Pan, Like Lin, Yineng Huang, Linglu Wu, Sitong Bao, Haiming Lu, and Yidong Xia, Robust half-metallicity in non-metal atoms intercalated two-dimensional GaN bilayer, Appl. Phys. Lett., 123, 042106 (2023). (https://doi.org/10.1063/5.0156210) 感谢陆海鸣老师课题组供稿!
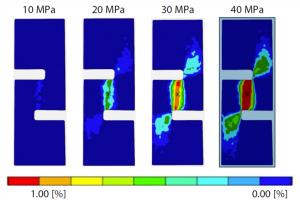
利用均质化分析验证改进 GFRP 平纹编织材料缺口压缩层间剪切试验方法的适用性
概述 近年来,玻璃纤维增强塑料(GFRP)因其高比强度、优异的可加工性和可塑性,已被广泛应用于电子基板、船舶和汽车外饰零部件等领域,成为不可或缺的材料。GFRP 是一种具有各向异性的复合材料,根据施加应力的主轴方向表现出复杂的变形和断裂行为:拉伸、压缩、弯曲、面内剪切、层间和面外剪切或这些行为的组合。 使用计算机辅助工程(CAE)设计产品时,迫切需要能够单独评估各部件失效行为的测试方法。而在缺口压缩层间剪切强度试验中,层叠方向的试验片尺寸短至 3.5~6.5 mm。由于层间剪切应变集中区域很窄,无法获得剪切应变。本项目研究了改进的缺口压缩测试方法是否适用于平纹编织的 GFRP,通过数值模拟重现弯曲、剥离等受基体树脂特性影响较大的变形行为,准确掌握层间剪切特性。 亮点 根据 X 射线 CT 细观结构观察结果建立 CAE 分析模型从摩擦系数的角度针对试件的约束条件进行均质化分析测量值与分析结果吻合良好 准备参数 GFRP 平纹编织材料层间剪切性能评估 试样为厚度约 10 mm 的 GFRP 平纹编织平板,缺口槽间隙宽度设计为 1 mm。依次涂抹黑色和白色喷剂,形成随机图案样式。 图1:试样形状 采用岛津精密万能试验机 Autograph AGX-50kNV、非接触式引伸计 TRViewX 和 GOM Correlate 软件获得平纹编织 GFRP 的层间剪切应力-应变关系图。在剪切应力达到 30 MPa 前为线性关系,之后呈现非线性关系,50 MPa 左右屈服。由 0.1 ~ 0.3 % 应变与应力的关系,通过最小二乘法计算得到的层间剪切模量为 2546.1 MPa。在 10 ~ 40 MPa 下 DIC […]
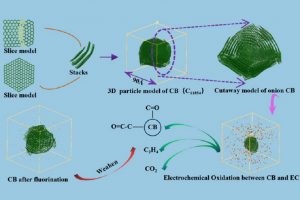
【中南大学肖劲-仲奇凡教授课题组】锂离子电池用导电炭黑微观结构建模及基于ReaxFF与DFT的电化学反应机理研究(Energy & Fuels 2023)
摘要 中南大学肖劲-仲奇凡课题组,通过高分辨率透射电子显微镜(HRTEM)、XRD、拉曼光谱和XPS实验检测,探讨了导电炭黑的结构特征。条纹的CB晶格长度大多<20Å,峰值约为10Å。条纹在0–360°处均匀分布,符合其“洋葱状”结构。总共堆叠了2-5层,平均堆叠数量为2.91,具有一定的顺序。弯曲条纹总长度的比例为67.13%,高于总数的比例(46.57%),表明弯曲条纹一般较长。然后通过构建CB(C11854)的洋葱状颗粒模型,结合FT-IR和XRD光谱计算验证了该模型的合理性。 作者同时在理论方面,采用基于反应力场的分子动力学(AMS软件ReaxFF模块)研究了CB和碳酸亚乙酯(EC)在锂离子电池中的电化学氧化行为。EC通过环内和环外O攻击CB,EC的分解产生CO2和C2H4。CB表面形成了各种O基团,EC和CB的破坏影响了电池的循环稳定性和寿命。在使用F2作为CB的保护后,EC的消耗减少,并且在CB表面仅形成少数O基团。这一结果为减缓CB和EC之间的电化学副反应提供了高精度的模拟支持。 扩展阅读: 中文详细解读:https://mp.weixin.qq.com/s/df7GchOCPdT_kVCdgKBNZw英文文献全文:Construction of a 3D “Onion-like” Model of Conductive Carbon Black for Lithium-Ion Batteries and Exploration of the Electrochemical Oxidation Mechanism of CB and Ethylene Carbonate via ReaxFF MD, Energy & Fuels. 2023, 37, 9, 6778–6790

I-FIT沥青混凝土的微观力学建模
概述 柔性路面常用分析和设计方法的主要局限之一是缺乏适当的材料表征,即沥青混凝土(AC)的粘弹性、粘塑性、各向异性;颗粒材料的应力相关、各向异性;以及通过结构能力分析预测开裂导致的基底断裂响应和各种形式的破坏。AC 材料的应变响应会受到 AC 混合料微观结构特征的影响,如骨料的尺寸和分布、基体性能等。尽管实验和连续介质断裂模型可以通过计算断裂能、应力强度因子(SIF)或其他参数理解均质化响应,但并没有区分在裂纹尖端能够发挥显著作用的微观结构特征。 本研究使用微观力学有限元模型研究 AC 在半圆弯曲(SCB)断裂试验、伊利诺伊州柔韧性指数试验(I-FIT)过程中的行为;利用数字图像相关(DIC)技术计算的应变场及测量的全尺度应力对模型进行多步验证;建立微观力学模型评估骨料级配、骨料分布和空隙空间等微观结构特征对 AC 断裂行为的影响。 有限元模型 两个二维有限元模型(FEM):线弹性均质模型和粘弹性非均质微观力学模型,都通过平面应变有限元简化二维空间中的采样。 线弹性均质模型 遵循 AASHTO TP 124 在 ABAQUS 中表示荷载和几何形状。二维 I-FIT 模型如所示,直径为 150 mm,在中心处锯开一个长 15 mm、宽 1.5 mm 的缺口。采用平面应变假设,厚度为 50 mm。试件由两根相对于切口中心对称放置的无摩擦杆支撑,以 50 mm/min 的速率施加位移控制载荷。关于网格配置,缺口周围采用三角形平面应变单元,其余部分采用四边形单元。 图1:低密度聚乙烯(LDPE)试样(a)试验(b)FE模型 粘弹性非均质微观力学模型 对于粘弹性微观力学模型,由骨料和砂浆组成。假设骨料相为线弹性,弹性模量为 60 GPa;砂浆相定义为沥青结合料、空隙和过 2.36 mm 筛骨料的组合,设定为线性粘弹性。 采用两种方法定义 I-FIT 试样中的骨料分布:(1)基于实际 I-FIT 试样测试前获得的图像数据,导入 Simpleware 软件分割出不同的相;(2)基于 Python 脚本在 I-FIT 几何形状上创建随机骨料分布,与特定配料设计具有一致的骨料级配和体积比。 图2:骨料分布试样(a)DIC 和 […]

经导管主动脉瓣植入术中支架放置和旋转方向对假体小叶内应力的影响
概述 主动脉瓣狭窄(AS)是一种瓣膜小叶钙化和结构扭曲逐渐抑制正常功能的疾病。AS 的常规治疗是外科瓣膜置换术(SVR),但因其极具侵入性,大约 31.8% 的患者被认为不适合该手术。因此,开发出一种替代的微创治疗方案即经导管主动脉瓣植入术(TAVI)。 TAVI 装置是个圆柱形支架,但在植入后会由于原生阀瓣上的钙化材料对支架施加不规则力引起局部膨胀。因此,变形TAVI装置内的小叶可能会承受增大的应力,导致装置过早失效。本项目通过计算分析,模拟一个完整的 TAVI 装置模型,并将其与由 CT 数据获得的主动脉根模型整合,随后进行压力模拟的心动周期。 亮点 在 Simpleware 软件中处理患者 CT 扫描数据,分割主动脉根部并与 TAVI 装置整合;在 ABAQUS 中模拟该装置的心动周期;通过将假体放置在不同角度方向的模拟评估对小叶应力的影响。 方法 获取 83 岁患者心脏舒张期的 CT 扫描数据,导入 Simpleware 软件进行图像处理,分割出主动脉根部、与退行性主动脉狭窄相关的小叶和 8 个钙化肿块。将基于 SAPIEN XT 的 TAVI 装置与主动脉根部整合,并生成高质量的网格模型。 图1:钙化肿块、主动脉根壁和主动脉小叶 然后在 ABAQUS 中模拟该装置的心动周期,重复 8 次,每次装置都相对原生瓣膜处于不同的旋转方向。装置的方向由原生小叶与假体小叶间的夹角定义,模拟的角度 θ 分别为0°、15°、30°、45°、60°、75°、90°和105°。 图2:TAVI 装置小叶(蓝色)相对于原生小叶(黑色)的旋转角度(绿色) 图3:完整 SAPIEN XT 和 NovaFlexþ 输送装置的计算模型 主动脉根部、主动脉、左心室流出道(LVOT)和原生小叶的密度为 1.1 g/cm3,瑞利阻尼系数为 α=800(β=0)。假设钙化肿块的密度为 2g […]