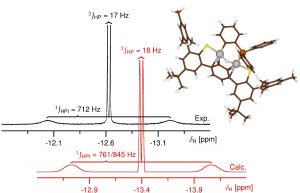
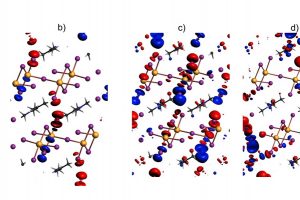
卤化铅钙钛矿,如(CH3NH3)PbI3,其优异的半导体性能以及在太阳能电池、光催化、LED中的成功应用,使其成为近期广泛研究的重点材料。 层状钙钛矿,如(C4H9NH3)PbI稳定性更好,同时支持更大的功能化有机阳离子,又能保持其母体钙钛矿的良好半导体特性。但铅化合物具有毒性,这促使科学家们寻找其他卤素金属化合物作为替代品。 碘化铋化合物似乎很有前途,因为铋化合物通常没有毒性,如包含双核阴离子Bi2I93-的(CH3NH3)3Bi2I9,在光伏器件上进行了测试,但经过大量优化后,效率只有3.17%。相比之下,卤化铅钙钛矿太阳能电池目前能达到20%以上。造成这一差异,结构化学方面有一个重要原因:碘化铅易形成层状或网格阴离子,而铋主要形成分子或链状阴离子。迄今为止,只有两种层状碘化铋阴离子化合物被报道( Chem. Mater., 2015, 27, 7137–7148;Inorg. Chem., 2000, 39, 6107–6113)。从层状混合卤化铋化合物(TMP)1.5[Bi2I7Cl2](TMP = N, N, N’, N’ – tetramethyl-piperazine)的合成来看,只要能找到合适的相反电荷离子,就可能合成更多的有机-无机层状碘化铋化合物。 马堡大学Ralf Tonner与Johanna Heine等人,报道了层状有机- 无机碘化铋化合物(Me2C=NMe2)Bi2I7,这是第一个这类层状化合物,金属位置实现完全占据。对晶体的合成、反应活性、晶体结构和光学特性进行了研究,揭示其独特的拓扑结构,高稳定性和低带隙特性。用量子化学方法分析了化合物的电子性质,提出对扩展固体中碘-碘接触的表征以及定量化的方法。文中还讨论了为什么该发现与卤化物钙钛矿铅有关,以及如何简易地制备离子,为金属卤化物材料开辟了新的机会。 作者使用VASP对晶体结构进行优化,使用AMS中的BAND模块进行周期性体系的能量分解分析、成键机理研究(pEDA-NOCV)以及QTAIM分析。pEDA的分析结果令人惊讶,这些双层之间的相互作用能53%来自色散能,47%来自电子,这表明结合机理比纯范德华结合更为复杂。进一步研究pEDA中的“吸引力”项(总结合能中的负值项)表明,静电相互作用占主导地位(56%),这可以理解为有机层和无机层的离子性导致。然而轨道相互作用占44%,这表明共价作用的贡献也非常显著。 通过化学价自然轨道方法(NOCV)进一步分析这些轨道相互作用,确定共价作用主要来自于碘-碘之间的电荷转移。 参考文献: Natalie Dehnhardt, Jan-Niclas Luy, Marvin Szabo, Mirco Wende, Ralf Tonner and Johanna Heine, Synthesis of a two-dimensional organic–inorganic bismuth iodide metalate through in situ formation of iminium cations, Chem. […]