从AMS2020开始,AMS诸多功能由AMS驱动各个模块完成,例如频率计算,算法由AMS实现,可以使用该算法调用不同模块完成频率计算。其他功能,例如NEB过渡态搜索、巨正则系综蒙特卡洛模拟、Force Bias蒙特卡洛模拟、分子动力学模拟、势能面扫描等也是如此。因此2021版以及以后的版本,将在驱动算法、ADF等各个计算引擎两方面均会有所改进。 AMS驱动 自动反应路径搜索:AMS驱动中的新功能PES exploration,在调用不同计算引擎时,能够自动帮助用户发现过渡态和局部极小值 Process search:找到极小值和连接它们的过渡态 Saddle search:寻找附近的过渡态 Basin hopping:寻找局部极小值 Landscape refinement:支持用户使用不同的理论,精修先前定位的过渡态和能量极小值点 Binding Site:确定并可视化簇或表面上的结合位点 PES Exploration相关教程:hydrohalogenation、water dissociation on ZnO(10-10). 添加球形电位墙:该势在墙内为0,在墙外存在向内推的力,具体使用方法与势的形式,参考说明书 D4色散修正,从仅仅支持ADF扩展到支持ADF、BAND、DFTB force bias Monte Carlo:对所有模块均支持(AMSinput – Model – force bias MC (fbMC) ) 力场:ForceField的速度提高了2-3个数量级 ParAMS:用于拟合DFTB、ReaxFF参数 ADF 极化力场:QM/FQ Quantum Mechanics/Fluctuating Charges 非弛豫偶极矩激发态 激发态间跃迁偶极矩 快速激发态、吸收光谱计算 – POLTDDFT:增加了大量元素,适合大多数元素 r2SCAN-D4 泛函 Eigenvalue-only self-consistent GW (evGW) 适用于多体微扰理论的新TZ3P和QZ6P基组 新的配体场DFT(LFDFT)光谱计算:ESR g-tensor doublets, XMCD BAND r2SCAN-D4 […]

全溶剂化染料敏化TiO2光阳极中的光致电子注入
染料敏化太阳能电池(DSSC)和染料敏化光电化学电池(DSPECs)是近年来太阳能转换领域的研究热点。为了发挥这些器件的潜力,应该更清楚地了解它们的化学稳定性和效率。由于效率与光诱导电荷分离和电荷复合的关键过程密切相关,分子的能级和界面的能级需要最佳匹配。 计算研究提供了对这些基本过程的深入研究,并提出了设计原则,但考虑到系统的复杂性,必须在精度和计算成本之间找到一个很好的折衷方案。莱顿大学和阿姆斯特丹大学的研究人员使用了基于DFTB和Extended Hückel方法模拟光致电荷分离和电子从有机染料注入TiO2电极。 用SCC-DFTB(ti-org-0-1)分子动力学模拟了完全溶剂化染料敏化光阳极系统(如图),研究了核动力学和显式溶剂化对光诱导电子注入过程的影响。研究了不同扩芯萘二酰亚胺(NDI)基染料。对于可靠的HOMO和LUMO能量,利用ADF(B3LYP-D3-BJ/DZP,COSMO),使用delta-SCF和TDDFT计算,对Extended Hückel哈密顿参数进行了优化。用该方法计算的NDI基染料的氧化还原电位与实验值吻合较好,可用于合成未知物种。 核动力学和轨道取样,对描述注入过程很关键,而显式溶剂化对分子动力学过程中探索正确的构象空间非常重要。 参考文献: J. P. Menzel, A. Papadopoulos, J. Belić, H. J. M. de Groot, L. Visscher, F. Buda, Photoinduced Electron Injection in a Fully Solvated Dye-Sensitized Photoanode: A Dynamical Semiempirical Study, J. Phys. Chem. C 2020, 124, 51, 27965–27976.
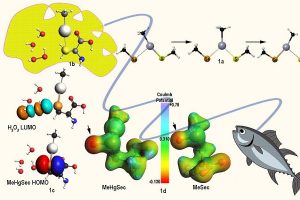
甲基汞中毒机理的第一性原理研究
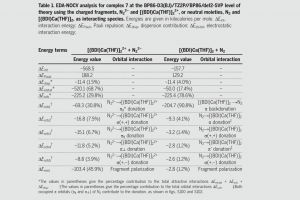
甲基汞是一种亲电毒物,被世界卫生组织列为十大主要公共卫生关注物质之一。与生物相关硫醇和硒醇结合、破坏它们的功能,被认为是其毒性的分子基础。然而原子级别的实验研究,例如通过X射线结晶学,相当难以开展。来自Università degli Studi di Padova (意大利) 和 Universidade Federal de Santa Maria (巴西)的学者们,利用AMS的相对论密度泛函理论方法,通过三个步骤研究了甲基汞化学,模拟了甲基汞与硫蛋白和硒蛋白的相互作用和反应性。 1) 使用配体交换反应来模拟结合步骤(图1a),其中目标硒酸盐攻击金属中心,导致载体硫醇盐的置换。能量分解分析(EDA)表明配体交换基本上是分散的SN2@Hg 反应。 2) 通过溶剂辅助质子交换模型研究了甲基汞结合后,硫核还原过氧化物的能力,从而理解甲基汞硫还原过氧化物的速率介于完全质子化和完全去质子化硫之间,产生相应的硫氧化物(图1b和1c)。 3) 最后,研究了金属成键促进硒碳键断裂的机理假设,发现金属将电荷移向硒氧化物的氧的能力,促进了β-消除反应伴随硒碳键断裂,从而导致靶硒蛋白的不可逆抑制(图1d)。 这幅图描绘了毒素从环境(右下)到生物目标(左上)的移动过程,生物目标为一个中毒(黄色)的大脑。 a) 亚硒酸甲汞络合物与硫醇盐的配体交换反应的反应物络合物、过渡态和产物络合物; b) 过氧化氢辅助少量水分子氧化半胱氨酸甲基汞络合物; c) 甲基汞配合物的HOMO和过氧化氢的LUMO,主要参与氧化过程; d) MeHgSec和甲基硒代半胱氨酸(MeSec)的分子静电势图显示,甲基汞结合后,硒功能的氧原子上有较大的负电荷积累。 参考文献: Madabeni, A., Dalla Tiezza, M., Omage, F. B., Nogara, P. A., Bortoli, M., Rocha, J. B. T., and Orian, L. Chalcogen-Mercury Bond Formation and Disruption […]
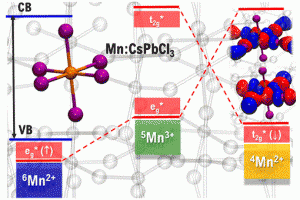
锰掺杂卤化铅钙钛矿:能量转移机制还是电荷转移机制?(ACS Energy Lett. 2021)
Mn掺杂的铅卤化铅钙钛矿的掺杂发光寿命长,主体激子量子产率高。沙特阿拉伯国王大学Edoardo Mosconi课题组与意大利技术研究院Filippo De Angelis课题组等,通过对APbX3钙钛矿(X=Cl,Br,I)的DFT计算,研究了Mn掺杂钙钛矿敏化掺杂发光过程中,能量和电荷转移的争议问题。 作者定量地模拟了Mn掺杂钙钛矿在不同电荷和自旋状态下的电子结构,将Mn敏化作为钙钛矿组分的函数,对结构/机理进行了分析。该分析的结果,同时支持能量转移机制和电荷转移机制。后者可能更适合于Mn:CsPbCl3,因为它具有较小的能量势垒,并规避了自旋和轨道方面的限制。在电荷转移的情况下,决定掺杂发光量子产率的一个重要因素是中间氧化物种的能量,而带隙共振可以很好地解释能量转移。这两个方面由钙钛矿主体的带边能量控制,而这又可以被卤化物X所调制。 参考文献: Damiano Ricciarelli, Daniele Meggiolaro, Paola Belanzoni, Asma A. Alothman, Edoardo Mosconi*, and Filippo De Angelis*, Energy vs Charge Transfer in Manganese-Doped Lead Halide Perovskites, ACS Energy Lett. 2021, 6, XXX, 1869–1878
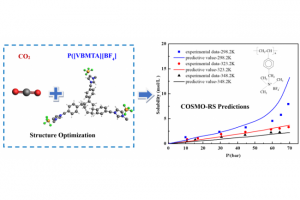
基于COSMO的气体在聚合物离子液体中溶解度的预测模型(Green Energy & Environment 2021)
气体在聚合物中的溶解度在加工先进高分子材料中起着重要的作用,聚合物离子液体(PILs)是一类重要的、尚未开发的聚合物。北京化工大学雷志刚教授课题组,最近在Green Energy & Environment的论文中,将不同的COSMO-RS参数与AMS软件COSMO-RS模块中的聚合物功能相结合,预测气体在标准聚合物和PIL中的溶解度。对于常规聚合物,标准COSMO-RS参数化性能最好,而对于PIL中的CO2溶解度,离子液体参数(ADF Lei 2018,见下文)的预测最为准确。 下图为COSMO-RS (ADF 2018 Lei) 预测CO2在聚合物离子液体 P([VBMTA][BF4]) 中的溶解度结果: 2018年雷志刚教授在Green Energy & Environment发表的一篇文章中,基于ADF框架对离子液体COSMO-RS参数的进行了重新参数化。训练过程使用了包括2283个无限稀释活度系数数据点和1433个从文献中收集的CO2溶解度数据点。并利用修正后的参数预测了CO2在低温(<273.2k)下在纯离子液体中的溶解度,以及CO2在宽温压范围内在混合离子液体中的溶解度。 如何使用该参数? 在ADF的COSMO-RS模块图形界面,用户可以直接选择Method > Parameters > ADF Lei 2018: 参考文献 Ruisong Zhu, Zhigang Lei, COSMO-based models for predicting the gas solubility in polymers, Green Energy & Environment (2021) Jingli Han, Chengna Dai, Gangqiang Yu and Zhigang Lei, Parameterization of […]
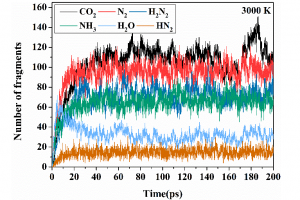
采用理论模拟、在线光电离质谱和热重-DSC-红外-质谱揭示N-脒基脲二硝酰胺盐的热解机理(Combustion and Flame 2021)
N-脒基脲二硝酰胺盐(GUDN)由于高能量和低感度受到了研究者的关注。含能材料的热分解动力学行为研究对其实际应用具有重要的作用。然而,在实验技术上确认反应活性中间体仍然是有待解决的问题。本文作者通过ReaxFF反应分子动力学联合在线光电离质谱和热重-DSC-红外-质谱阐明了GUDN的热分解机理,构建了GUDN热分解网络。结果表明:GUDN热分解的主产物包括CO2、N2、H2N2和NH3,少量产物包括H2O和HN2,产物NO2,NO和CO仅参与初始生成和中间体转化反应。本研究可为探索热动力学参数和进一步建立点火模型提供一定的理论基础。 此项研究中分子动力学模拟在AMS(Amsterdam Modeling Suite,原ADF)软件ReaxFF模块中完成。 参考 原文:Liping Jiang, Xiaolong Fu, Xuezhong Fan, et al. Combustion and Flame. 2021, 229, 111406. 案例:ReaxFF-燃烧:甲烷燃烧过程模拟https://www.fermitech.com.cn/wiki/doku.php?id=adf:simplemdofreaxams 本文由西安近代化学研究所姜丽萍博士、付小龙研究员供稿。
低价钙中氮的络合与还原(Science 2021)
最近埃尔朗根-纽伦堡大学S. Harder课题组、马尔堡菲利普大学G. Frenking课题组、南京工业大学赵莉莉教授课题组等,报道了在氮气(N2)下以LCa-CaL(其中L为β—酮二亚胺配体)的形式制备低价CaI络合物的尝试,使LCa(N2)CaL分离,并对其进行晶体学表征。 该络合物中的N22-阴离子,在大多数情况下可作为一种非常有效的双电子供体。因此该研究的目标是将LCa(N2)CaL作为低价CaI络合物LCa-CaL的合成子。N22-阴离子也可以质子化为二氮烯(N2H2),然后歧化为肼和N2。研究组讨论了Ca d轨道在N2活化中的作用。 本文与赵老师2018年在Science上的文章 Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr, or Ba) that mimic transition metals, Science 361, 912–916 (2018)一样,使用了AMS中ADF的EDA-NOCV功能,分析络合成键中金属d轨道的作用,并对轨道作用能、泡利排斥能、静电作用能进行了分析。 参考文献 Dinitrogen complexation and reduction at low-valent calcium, Rösch et al., Science 371, 1125–1128 (2021) 相关教程 EDA-NOCV功能案例(配位键作用):配合物的EDA-NOCV分析(Science 2018)
AMS在化工分离过程中的应用
概述 COnductor-like Screening MOdel for Realistic Solvents(COSMO-RS、COSMO-SAC),是一种基于DFT数据,预测气体纯液体、液体混合物、溶液、离子液体性质的方法,也用于共晶的研究。数据库中包括2500种化合物(溶剂、小分子)以及离子液体离子的DFT数据库,用户也可以通过ADF模块轻松扩充DFT数据库。 除了基于DFT数据的COSMO-RS、COSMO-SAC方法,还有基于基团特性的UNIFAC方法。用户提供分子结构SMILES,程序即可预测材料性质,甚至有时候能够得到更好的结果。 可以预测的流体性质列表 溶解度、分配系数 (log P, log Kow)pKa、pKb、Sigma Profile活度系数、溶剂化自由能、亨利常数饱和蒸汽压、沸点、(二元/三元)气液平衡相图 (VLE/LLE)过剩能, 共沸、溶混性gap成分线、混合物闪点对单组份物质的熔点、沸点、闪点、临界温度等性质的快速预测 溶剂筛选方面的应用(参考教程) 化学工程师可能希望通过调整蒸气压和沸点来优化蒸馏过程,而药物化学家可能希望最大限度地提高溶剂萃取效率,将药物活性成分从主要污染物中分离出来,或者最大限度地提高辅料的溶解度。 我们的溶剂系统优化工具,能对一组给定溶剂,为溶液、液-液萃取寻找最佳溶剂混合物,从而完美减少实验搜索空间。脚本工具可以方便地根据指定特性,筛选溶剂或溶剂组合。 COSMO-RS/SAC的脚本应用还包括优化催化剂、溶解度参数、pKa值和在离子液体中的吸附。 聚合物方面的应用(参考教程) COSMO-RS和COSMO-SAC能够预测聚合物的热力学性质和描述符,如活度系数、蒸汽压、分配系数、溶解度和Flory-Huggins-Chi 应用实例 实例1:双相萃取5-羟甲基糠醛溶剂筛选的多尺度模拟与实验研究 Zhaoxing Wang, Souryadeep Bhattacharyya and Dionisios G. Vlachos Solvent selection for biphasic extraction of 5-hydroxymethylfurfural via multiscale modeling and experiments Green Chem., 2020,22, 8699-8712 可再生的木质纤维素生物质衍生平台化学品,如糠醛和5-羟甲基糠醛(HMF),为未来的聚合物、精细化学品和药物提供了非常有希望的合成途径。副反应严重抑制水相中果糖脱水产生的HMF产量,但通过将HMF从反应水相中提取到有机相中可以将副反应降至最低。不过理想溶剂的筛选是非常重要的。 为了提高溶剂筛选效率,特拉华大学的研究人员最近将AMS软件中COSMO-RS理论预测与靶向实验相结合,研究果糖脱水反应中HMF的提取。使用COSMO-RS中的ADFCRS-2018数据库,首次预测了298 K和423 K(代表性木质纤维素生物质反应温度)下所有潜在“水-溶剂”对的“水-有机”液-液平衡。当存在混溶间隙时,利用平衡相组成,对果糖脱水反应中遇到的HMF、乙酰丙酸(LA)和甲酸(FA)的logP初始化计算。 实验测量了多个同系物系列溶剂的分配系数,以评价COSMO-RS预测的准确性。典型误差因子在2以内,表明该方法非常适合于筛选。首次在原位测定了这些物种的高温分配系数。这项研究确定了新型高效萃取剂,如取代胺和苯酚,用于HMF的反应萃取。经过实验验证,其分配系数比传统萃取剂提高了一个数量级。其他标准,如溶剂热稳定性,反应性和毒性,也进行了评估,更多细节,请参考原文。 实例2:人工智能和热力学协助解决纵火案 Sander Korver, Eva Schouten, […]

AMS在石油化学工业中的应用
概述 随着计算机性能与计算理论方法的改进,在原子分子水平模拟分子的结构与行为,在石油化工领域得到越来越广泛的应用。在高分子材料、分子筛催化剂以及油品添加剂的研发中,计算模拟能够帮助研究者更深入地理解所研究的体系,促进新分子筛催化剂、高分子材料的的开发与改性,油品添加剂新产品研制,减少实验工作,缩短研发周期。 分子筛催化 结构 分子筛存在大量的真空区域,基于平面波的DFT计算将耗费大量时间在无意义的真空区域。BAND采用STO+NO基组,从而大大节省计算时间,使用GGA等泛函进行结构优化具有较好的可行性 吸附与扩散 BAND、DFTB提供D3(BJ)、D4(EEQ)色散修正,更精确的计算分子吸附 自动探索表面吸附位点,自动探索表面化学反应机理 通过基于DFT、MOPAC、DFTB或力场的分子动力学模拟能够非常便捷地计算扩散系数 分子筛与分子之间相互作用能量的分解分析,以及电子转移的定性图像 巨正则系综蒙特卡洛、分子动力学模拟MOF材料表面的吸附等温线 高分子催化 高分子聚合催化剂的结构优化 高效并行的ADF能够支持大分子的DFT结构优化 支持多尺度方法,用户可以灵活划定聚合物原子区域,对不同区域采用不同的理论,协同优化聚合催化剂结构 对于一维周期性结构BAND采用的STO+NO基组大大提高计算效率,能够以比相对于平面波方法低一个数量级以上的计算成本,完成高精度GGA结构优化 高分子催化反应机理 吸附结构优化 多尺度方法计算反应过渡态、活化能、反应历程 对于一维周期性结构,支持高精度、高效率的DFT过渡态搜索、活化能计算、反应历程 使用ReaxFF分子动力学模拟对聚合物的力学性质,如杨氏模量、屈服点、泊松比进行预测 ReaxFF分子动力学探索未知反应机理 根据反应物、产物的分子结构,筛选反应通道 快速确认(副)反应可能性、(副)产物可能性 确定化学反应中反应物和产物之间的最佳原子映射 聚合物与分子之间相互作用能量的分解分析,以及电子转移的定性图像 油品添加剂 热解与燃烧 DFTB、MOPAC、ReaxFF均可作为分子动力学模拟引擎,支持同样的结果分析工具;ReaxFF作为经典的热解与燃烧分子模拟工具,包含最丰富的反应力场,广泛应用于该领域的研究 Bond Boost、REMD、CVHD、fbMC等加速反应方法,能够让分子动力学模拟在实 验温度下,得到宏观时间尺度化学反应的结果 Mol Sink允许在分子动力学模拟过程中,定时清除指定产物;Mol Gun可以定时添加反应物 自动分析产物数量变化、基元反应、反应速率常数,评估 其他研究工具 COSMO-RS 气-液相平衡、液-液相平衡 最优萃取溶剂优化 活度系数、溶解度、pKa、logP 使用定量构效关系估算物质性质:密度、熔点、沸点、闪点、介电常数、液态摩尔体积、分子范德华体积与表面积等 EDA-NOCV 键能分解分析,对分子间范德华作用、氢键作用能进行分解分析,分析泡利排斥、静电作用、轨道作用、色散作用能大小 分析化学键形成机理,电子在分子轨道中的定量转移,以及定性图像 固体表面与分子之间相互作用能量的分解分析,以及电子转移的定性图像 分子动力学 粘度 研究实例 实例1:Co/Mn/Na/S催化高选择性费托反应 Jingxiu Xie, Pasi P. Paalanen, […]
AMS在有机发光显示材料中的应用
概述 由于智能手机电子屏等巨大市场规模,OLED材料和有机电子学是一个非常活跃的研究和工业发展领域。有机发光显示材料的研究是一个综合性的系统工程问题,有分子层面的问题,也有工艺等等层面的问题。在分子层面,电子的激发形成激子、载流子的迁移、分子激发态的内部转换以及发光过程,目前已经有相当完善的理论研究方法和工具。 ADF模块具有一些独特的工具来模拟这些分子水平上的过程问题,如电荷传输、激子耦合和发光效率等。对优化有机电致发光二极管(OLED)、有机场效应晶体管(OFET)、光伏电池(PV和OPV)和染料敏化太阳能电池(DSSC)等有机电子器件中的材料性能而言,这些分子过程非常重要。 AMS提供的模型和工具 材料结构与电子结构 通过ZORA、X2C方法精确考虑相对论自旋轨道耦合效应 包含最新的色散修正泛函,以及高精度泛函,准确预测分子结构以及分子间作用 使用高精度STO/NO基,优化分子晶体结构 分子能级与轨道 带电场、溶剂环境的表面-分子相互作用 大体系高效激发态计算 单重激发态能量Sn 三重激发态能量Tn 自然跃迁轨道NTO ΔEST、重整能 激发态结构优化与频率计算 势能面最低交叉点MECP 磷光、荧光辐射跃迁寿命 聚集诱导发光 包括QMMM在内,DFT、半经验量子化学方法、力场的多尺度方法 考虑色散修正的分子晶体、团簇结构优化 多尺度方法TDDFT进行Sn、Tn激发态计算 多尺度方法的激发态振动频率计算 分子间载流子迁移速率、转移积分 激发态动力学 自旋轨道耦合矩阵元SOCME,ZORA、X2C方法更加精确、高效 Franck-Condon因子、Huang–Rhys因子,及其多尺度计算 分析转动、振动模式对Huang–Rhys因子、系间窜跃、内转换与发光的影响 (逆向)系间窜跃速率、内转换速率、辐射跃迁速率 发光量子产率PLQY、外部量子效率EQE AMS发展展望 2020年起,荷兰SCM公司与Simbeyond公司合作开发第一款完全集成的OLED多尺度仿真平台,结合两家荷兰公司在工业有机器件和原子级学术模拟中的优势,实现跨越分子到器件的工业研究, 目前已经开发出OLED材料数据库与流程化模拟脚本。(相关技术资料2022版) 其他参考资料: 有机电子学模拟讲义I 有机电子学模拟讲义II OLED相关墙报 有机电子学领域文章合集 特邀文集 中文教程库 立即试用AMS http://www.scm.com/free-trial 研究实例 实例1:无重元素高效持久室温磷光分子 Indranil Bhattacharjee, Shuzo Hirata Highly Efficient Persistent Room‐Temperature Phosphorescence from Heavy Atom‐Free […]