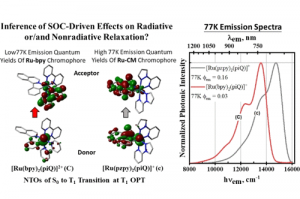
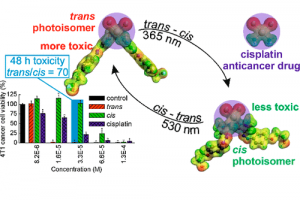
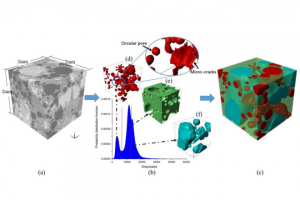
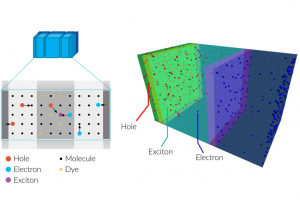
电荷转移态中的自旋轨道耦合 阿姆斯特丹大学的研究人员研究了电子供-受体分子的扭曲对性质的影响。对于包含芘受体和二甲基苯胺供体的电子供体-受体体系,确定了自旋轨道耦合矩阵元(SOCME)、电荷分离的电子耦合,对构象的依赖性。 对热激活延迟荧光(TADF)、光动力疗法、三重态发光二极管而言,自旋轨道耦合效应起着决定性影响。作者在动力学和能量角度,讨论了旋-轨电荷转移系间窜跃 (SOCT-ISC) 机制,包括经典Marcus电子转移理论中,电荷分离、电荷复合的相关参数。自旋轨道耦合,在电荷复合到三重态过程中起着重要作用,可以通过TD-DFT 进行探索,同时TD-DFT也为理解和预测 SOCT-ISC 机制提供了有效途径。该研究用丙酮和 4-硫代胸腺嘧啶的自旋轨道耦合矩阵元作为基准。 关于这项工作的三个报告的视频资料,可以辅助读者理解如何在自己的工作中使用类似的方法: 芘-二甲基苯胺正交电荷转移态的自旋-轨道耦合_René Williams 使用计算化学来描述和理解SOCT-ISC机制_Davita van Raamsdonk 正交电荷转移态中的自旋轨道耦合_Shivan Bissesar 所有 ADF 输入文件(链接)。 Bissesar, D. M. E. van Raamsdonk, D. J. Gibbons, R. M. Williams, Spin Orbit Coupling in Orthogonal Charge Transfer States: (TD-)DFT of Pyrene-Dimethylaniline. Molecules 2022, 27 (3), 891. 热激子基 TADF 分子设计的理论探讨 近年来TADF过程的研究取得了诸多突破,但要进行更高效率和量子产率的TADF分子设计,还需要更多深入的理论机理上的研究。与传统(冷)TADF一样,基于热激子的TADF材料也可以有效地利用单重态和三重态激子,理论上产生100%的IQE。与冷TADF(从低激发T1到S1)不同,热TADF中的RISC过程发生在高激发三、单重激发态(Tm(m>1)与Sn(n>1))之间。设计满足热激子形成条件的材料,例如低三重态之间能隙足够大,而高激发单-三重态能级间隙足够小,仍然是相当困难的。 印度SRM大学化学系Jesni M Jacob、Mahesh […]